
Introduction
Rheumatoid vasculitis (RV), a severe systemic complication of rheumatoid arthritis (RA), is driven by multifactorial mechanisms linking autoimmune inflammation to vascular endothelial dysfunction. The study investigates the pathogenetic connections between hypercoagulability and systemic inflammatory responses in RA patients, emphasizing the pivotal roles of cytokines, immune complexes, and endothelial alterations. Elevated levels of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-17, alongside increased autoantibody profiles such as anti-citrullinated peptide antibodies (ACPA), underlie the immune-mediated damage to vascular walls. This damage is exacerbated by the deposition of circulating immune complexes, complement activation, and the recruitment of inflammatory cells, collectively leading to endothelial cell apoptosis, angiogenesis, and systemic hypercoagulable states [1]. At present, no single factor has been identified as solely responsible for inducing systemic rheumatoid vasculitis (SRV). This complication, as other extra articular manifestations of rheumatoid arthritis, has a multifactorial origin. It is believed that intimal and endothelial damage in SRV is associated with a combination of exogenous, immune and genetic specificities. Patients who had RA associated with p53 mutations were found to have a higher risk for SRV.
Another predisposing factor involves impaired immune tolerance, with the presence of HLA-DRB1*04/04 or HLA-C*03 alleles. Besides that, a connection between RV and KIR2DL3/HLA-C*0802 complex and cutaneous lesions were detected. Increased levels of macrophage migration inhibitory factor were found to enhance inflammation. Long standing RA, male sex, smoking, rheumatoid nodules and different alleles of HLA-I/HLA-II have been linked to an increased rate of RV. Other comorbidities predisposing to this type of connective tissue disease associated vasculitis are cerebrovascular and peripheral vascular disease, maybe due to already present vascular damage [2]. SRV is considered one of the most dangerous systemic complications of RA. The median age at presentation ranges from 62-68 years and the median duration of RA prior to SRV is 10-16 years [3]. Nowadays, the clinical prevalence is estimated from 1-5% (affecting predominantly one in 9 men and one in 38 women [4]), whereas autopsy studies reported 15-31% [1, 3]. It is strongly associated with smoking and advanced RA, as SRV was mainly found in patients with destructive, nodular, and long-standing RA.
Rheumatoid vasculitis usually affects medium and small size vessel, strongly resembling polyarteritis nodosa [5]. Currently, no definitive methods exist to distinguish SRV from other vasculitis, but a strong correlation between the presence of rheumatoid antibodies and SRV. In 90% of cases, patients with SRV are positive for ACPA (anti cyclic citrullinated peptide antibody), whereas only 40% test positive for ANA (antinuclear antibody) (showing a weaker correlation) [2, 6]. To better understand the importance of this study, we should assess the impact of SRV on the affected population. Its mortality rate is estimated to be around 30-50% and relapse in case of remission in 25% [7], while the treatment-related toxicity and disease complications often surpass these figures. This pathology involves skin (palpable purpura/nodules/ulcers/digital necrosis) [6] and peripheral nerve lesions (symmetric sensory polyneuropathy, mononeuritis multiplex) in more than 80% of patients. Rarely, major systems are affected: heart, kidneys and bowel, leading to complication including myocardial and bowel infarction as well as renal failure. These multi-organ manifestations are the main causes of death in SRV.
Material and methods
A search for scientific papers published since 1996 was conducted using the MEDLINE electronic database via the PubMed and HINARI (Health Internet Work Access to Research Initiative) search engines – part of the Research4Life program. Full-text articles available on these platforms were selected. The search terms used (in English) included: ”rheumatoid vasculitis”, ”rheumatoid arthritis”, ”hypercoagulability”, ”endothelial dysfunction”, ”cytokines”, ”immune complexes”, and ”systemic inflammation”. Original articles, meta-analyses, systematic reviews, and book chapters were included. No language restrictions were applied, but articles in English were prioritized. Additionally, the bibliographies of selected articles were reviewed to identify other relevant sources. The included studies focused on the mechanisms underlying the development of rheumatoid vasculitis.
Initially, the identification of relevant digital sources was based on a comprehensive review of the scientific literature, supplemented by other information sources, such as official reports and press articles. In total, 217 bibliographic sources were consulted and identified. The selection criteria included relevance to rheumatoid vasculitis, recent publication, and recognition in the scientific community.
Data on each identified pathogenic mechanism were collected from various sources, including official websites, scientific articles, and public information. Collected data included descriptions of mechanisms, functionalities, implementation methods, and reported outcomes.
Evaluation and comparison of the primary mechanisms and pathogenic factors were carried out using qualitative and quantitative methods. Each mechanism and factor was assessed based on its physiological and pathological characteristics, usability, data accuracy, and adaptability to different contexts. This process involved a thorough analysis of data and reported outcomes, comparing these against pre-established criteria relevant to the literature review's objectives.
The results were interpreted considering the specific requirements for addressing rheumatoid vasculitis, with a focus on its pathogenic factors (including cytokines, circulating immune complexes, and systemic inflammation). The study emphasized the methodological efficiency and effectiveness in managing endothelial dysfunction and hypercoagulability. Limitations and critical aspects of each pathogenic mechanism were evaluated in the context of practical needs and constraints.
Data validation was ensured by consulting and verifying information from multiple independent and reliable sources. This approach guaranteed the accuracy and reliability of the analyzed data, contributing to the study's validity and robustness.
Using these rigorous methods and diverse data sources, the study provided a detailed comparative analysis of the pathogenic mechanisms involved in rheumatoid arthritis that contribute to vascular damage. This facilitated the early identification and prediction of this severe systemic complication.
Results
Vascular endothelium relation with coagulation in inflammation
Endothelial cells have specific functions and adaptations aimed at preventing thrombus formation, maintaining blood fluidity and tissue perfusion. These functions are closely related to endotheliocytes activation and regulation of the extravasation of fluids, solutes, hormones, and macromolecules, as well as of platelets and blood cells during inflammation. There are two types of EC activation: Type 1, called stimulation and Type 2. Type 1 activation is independent of new gene expression, while Type 2 activation depends on gene transcription [8]. An example of Type 1activation is represented by inducing the histamine receptor. EC can be activated through GPCR (G-protein coupled receptor), for example by histamine coupling Histamine1-R (Gq). This process increases intracytosolic Ca2+ concentration, caused by PLC (Phospholipase C) activation. Further, this process stimulates PLA2 (Phospholipase A2) which cleaves phosphatidylcholine into arachidonic acid (substrate for Prostaglandin H2/Prostacyclin). Increased Ca2+ also activates NOS-3 (Nitric Oxide synthase 3). Besides that, Ca-calmodulin complex together with inhibited Myosin Light Chain Phosphatase determine increased contractility in actin filaments, part of tight junctions. This induces formation of intercellular gaps, which in addition to NO/Prostacyclin stimulates exudate formation. Using the same pathway, EC expresses both PAF (platelet activating factor) and P-selectin, providing tethering and activation of circulating neutrophils. Other adhesins, like CD99 (single chain type 1 glycoprotein) and CD31 (platelet-endothelial cell adhesion molecule 1) are essential for adherence and transmigration of monocytes and neutrophils. Type 2 EC activation is mediated by several cytokines. TNF-α (Tumour Necrosis Factor alfa) induces synthesis of E-selectin/VCAM1 (vascular cell adhesion molecule 1)/COX-1 and COX-2 (cyclooxygenase 1,2). Using the same pathway as TNF-α, IL-1 induces pro-inflammatory protein synthesis. These pathways typically act in a complementary manner, enhancing the inflammatory cell response. A specific feature for EC observed in chronic inflammation is the ability to present both MHC I and II (major histocompatibility complex I, II), thus activating T cells. It also stimulates angiogenesis by enhancing VEGF (vascular endothelial growth factor) transcription [9].
In addition, endothelial cells can secrete preformed vesicles (Weibel-Palade bodies) which store von Willebrand factor (VWF), P-selectin, and angiopoietin-2. These molecules are involved in platelet binding, leukocyte recruitment, and inflammation modulation. Furthermore, some endothelial receptors are involved in the generation of the important anticoagulant, activated protein C (aPC), which can interrupt the coagulation cascade by cleaving coagulation factors Va and VIIIa [10].
Pathophysiology of SRV
RA is an autoimmune disease, characterized by loss of tolerance to self-antigens, especially those containing citrulline residues generated by posttranslational modification. Coupling of Ab with Ag determines formation of IC (immune complexes). In addition, systemic inflammation determines a vascular reaction: increased permeability, upregulation of adhesion molecules, and enhanced expression of MMPs, PAD and pro-inflammatory cytokines. These adaptive immune responses determine formation of specific Ag (citrullinated/carbamylated proteins) which bind antibodies both at the site of inflammation and those in systemic circulation. Increased vascular permeability favours CICs (circulating immune complexes) deposition. Local IC formation and deposition determines progression and maintenance of inflammation through complement activation, coupling with specific receptor and activation of pro-inflammatory signalling pathways [11]. SRV, same as rheumatoid arthritis, has a multifactorial pathogenesis, with progressive discovery of new pathological linkages. The most important effectors are believed to be the immune complexes. Besides that, some cytokines tend to maintain and progress the inflammation. Immune and blood cells, through specific pathways (NET-osis, MET-osis, enzymes and cytokines secretion, and the release of cell-derived microparticles) induce and maintain the RV. Taken together, IC’s deposition, cytokine release and immune cell activation, determine blood vessel wall damage.
Effect on vascular endothelium
Immune complexes (ICs) play a critical role in the pathogenesis of any systemic immune mediated vasculitis, especially those affecting small blood vessels. RV is associated with small- and medium-vessel vasculitis in the context of connective tissue diseases. These types of angiitis mainly occur due to ICs deposition in the vascular endothelium, inducing or amplifies inflammation. Under normal physiological conditions, when the antigen-to-antibody ratio is close to 1, predominantly large ICs are formed. These are efficiently cleared through phagocytosis. In the condition of Ab excess, small ICs are formed, remaining in the serum and precipitating in the vascular bed, where they stimulate cytokine release and inflammatory pathways activation [12]. Once Ag-Ab complex transmigrated in subendothelial space, it will activate the inflammatory cells through Fc receptors (FcR). This will induce phagocytosis by activating ITAM (immunoreceptor tyrosine-based activation motif) and Cytokine transcription enhancement in all stimulated cells. Additionally, ICs bind to the C1q initiating the complement cascade, which determines chemotaxis (C3a, C5a), opsonization (C5b) and cytolysis (through membrane attack complex).
TNF-α, is a cytokine which mediates and promotes inflammation. There are two receptors responsible for its action: TNF-Receptor-1 (on nearly all cells) and TNF-Receptor-2 (on immune cells/heart). The binding of membrane-bound TNF-α to TNFR2 induces the expression of VEGFR2 via activation of endothelial tyrosine kinase (Etk). It also participates in activation of NF-kB (nuclear factor kB) by stimulating phosphorylation of IkBa (inhibitor of nuclear factor kB). This process facilitates translocation of the transcription factor in the nucleus. Besides stimulating NF-kB, TNFR1 also signals through p38MAPK (playing a role in rearrangement of cytoskeleton) and MAP3K/JNK (having a crucial role in pro-inflammatory molecules synthesis). Upon activation of the TNF-α–TNFR1 complex, JNK phosphorylates c-Jun and activating transcription factor-2 (ATF-2), further promoting inflammatory gene transcription. TNF-αAs a pro-inflammatory cytokine, TNF-α induces morpho-pathological changes in vascular endothelium. First of all, it provokes endothelial barrier dysfunction through several mechanisms. Activation of TNFR1 has been reported to induce p38MAPK/ERK pathway, which, through PKC-dependent rearrangement of the actin cytoskeleton, destabilizes microtubules and induces formation of intercellular gaps. It leads to an increased paracellular diffusion of plasma and macromolecules in endothelium. In parallel, TNF-α increases tyrosine phosphorylation of cadherin (activation of NF-kB) in vascular endothelium, decreasing intercellular adhesion. Secondly, TNF-α decreases NO levels (vasodilator) in endotheliocytes. Through TNFR1 stimulation, it suppresses the endothelial nitric oxide synthase (eNOS) gene promoter, inhibiting mRNA formation, consequently decreasing eNOS enzyme synthesis. Moreover, endogenous accumulation of ADMA (asymmetric dimethylarginine) directly inhibits eNOS by coupling to the active site. Thirdly, in a complementary manner, TNFR1 and TNFR2 stimulate the transcription of adhesion molecules (E-selectin/ICAM-1/VCAM-1) within 30-120 minutes of activation, mediating the recruitment and transmigration of circulating leukocytes into the vascular wall [13].
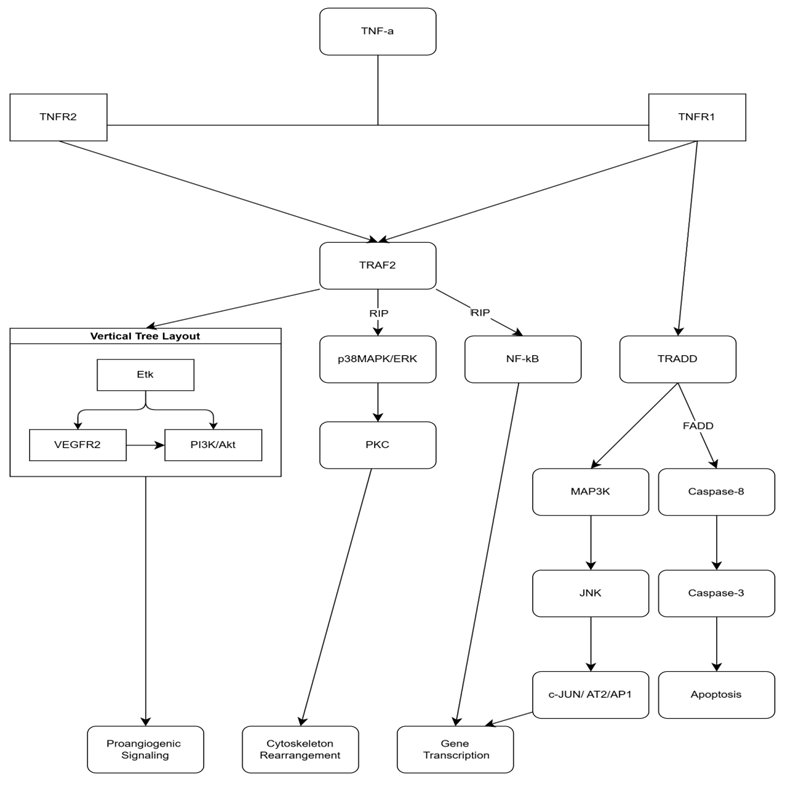 |
Fig. 1 Coupling both TNFR1/ TNFR2, TNF-α induces activation of TRAF2, except TNFR1-TNF-α complex activates TRADD-pathway too. FADD and RIP are playing the role of co-stimulators, inducing and enhancing signals. These cascade-like processes, mainly stimulate gene transcription, cytoskeletal rearrangements and angiogenesis or apoptosis. Note: TNF-α - Tumor Necrosis Factor-alpha; TNFR1 - Tumor Necrosis Factor Receptor 1; TNFR2 - Tumor Necrosis Factor Receptor 2; TRAF2 - TNF Receptor-Associated Factor 2; RIP - Receptor-Interacting Protein; NF-κB - Nuclear Factor-kappa B; TRADD - TNF Receptor-Associated Death Domain; FADD - Fas-Associated Protein with Death Domain; MAP3K - Mitogen-Activated Protein Kinase Kinase Kinase; JNK - c-Jun N-terminal Kinase; Caspase-8 - Cysteine-aspartic Protease 8; Caspase-3 - Cysteine-aspartic Protease 3; PI3K/Akt - Phosphoinositide 3-Kinase/Protein Kinase B; VEGFR2 - Vascular Endothelial Growth Factor Receptor 2; PKC - Protein Kinase C; p38MAPK/ERK - p38 Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase; Etk - Erythroblast Transformation Specific Kinase; c-JUN/AT2/AP1 - c-JUN/Activator Protein 1. |
IL-6 is one of the main pro-inflammatory cytokines in rheumatoid arthritis manifesting dose-dependent effects. There are several cell types which secrete IL-6: lymphocytes, monocytes, fibroblast, osteoblasts, endotheliocytes and mesangial cells. Mainly, cells secreting IL-6 are activated through TLR-DAMP (toll-like receptor-damage associated molecular pattern) / PAMP (pathogen associated molecular pattern) complex formation. The signalling pathway of the TLRs and the expression of inflammatory cytokines (e.g., IL-1, TNFα, and IL-17) work as it determines activation of NF-kB, similar to TNF-α. The stimulation of TLRs, IL-1, IL-17 and TNF-α activates IkB, which determines recruitment of NF-kB to the promoter of IL-6 gene. Further interaction with CREB induces hyperacetylation. This permeative action determines IL-6 gene transcription. IL-6 also stimulates differentiation of naïve T cells into Th-17 cells by inducing STAT3 transcription. In RA this pathway is more prominent because of citrullinated proteins, having role of PAMPs/DAMPs [14]. In order to induce an effect, this interleukin needs the presence of glycoprotein130 (coupled with JAK-Janus Kinase) on the membrane of the cell, activating either classical (IL-6+IL-6R) or trans-signalling (IL-6+sIL-6R) pathways. Besides having membrane receptors, it can also bind to the soluble receptor sIL-6R, formed by enzyme cleavage in myeloid cells and hepatocytes. sIL-6R is generated through proteolytic cleavage by enzymes such as ADAM10 in myeloid cells and hepatocytes. Importantly, trans-signaling enables IL-6 to act on cells that lack membrane-bound IL-6R—such as endothelial cells—but express gp130. Formation of this complex determines the activation gp130 signal transducing molecule, inducing JAK in target cells. Further, MAPK/STAT3/ERK1,2/Akt enhances proinflammatory molecules transcription, resulting in osteoclasts activation, Lymphocytes B differentiation and proliferation, metalloproteinases / VEGF/monocytes chemoattractant protein-1 synthesis [11]. To be more specific, classical activation with membrane receptor poorly induces activation of STAT3 (Signal transducer and activator of transcription 3), while trans-signalling strongly stimulates this mechanism. The other transcription factors are stimulated equally. IL-6 also contributes to inflammation by inducing the expression of ICAM-1, CCL2/MCP-1, and CXCL8/IL-8, promoting monocyte and neutrophil recruitment and adhesion to endothelial cells (ECs) [15, 16]. Also, stimulating ADAM10 expression through IL-8 and GM-CSF, determines cleavage of EC membrane CD162, thus increasing circulating neutrophils [17]. On the other hand, induction of ICAM1 membrane expression in vascular EC determines leukocyte adhesion and transmigration by coupling with LFA (integrin) [18].
IL-17 is a family of cytokines (IL-17A to IL-17F) secreted for host protection, have a major pathogenetic role in autoimmune and inflammatory diseases. It is synthesized mainly by TH17 cells, together with IL-6, IL-8 and MMP. TH17 cell activation and subsequent IL-17 secretion is stimulated by IL-6, IL1 and TGF-β [19]. The biological activity of IL-17 is mediated through a group of receptors, IL-17RA to IL-17RE. The principal signalling pathway activated by the IL-17/IL-17R complex involves the adaptor protein ACT1, which stimulates downstream activation of NF-κB and MAPK (mitogen-activated protein kinase) pathways, enhancing pro-inflammatory cytokine transcription [20, 21]. IL-17 accelerates the inflammation in vascular endothelium by stimulating vWF secretion. Von Willebrand factor mediates platelet (PLT) aggregation and stabilization of factor VIII. Importantly, IL-17 determines its effect on vascular endothelium, mainly through inducing cell apoptosis. It can be stimulated through Caspase 3 and 9 activation or by inducing Bax pathway [22]. IL-17 induces secretion of pro-inflammatory cytokines (IL-6, GM-CSF, IL-8 and CXCL1). When combined with TNF-α, IL-17 significantly enhances the expression of adhesion molecules such as E-selectin and ICAM-1. This synergistic effect is supported by transcriptomic data: IL-17 alone stimulates the transcription of 45 genes, TNF-α alone stimulates 1,036 genes, but together they stimulate 10,873 genes [23]. IL-17 amplifies ADP-induces PLT activation. It also enhances vWF production by EC, mediating PLT adhesion. Another mechanism consists of activating TF and inhibiting CD-39/ Thrombomodulin [24].
CD-20 is a B-cell membrane protein which basically participates in controlled regulation of activating IgD B-Cell Receptors (IgD-BCR). Over the long term, CD-20 also participates in expression of several other membrane proteins: CD19, CD81, CD22, CD40 and IgM-BCR, which are part of IgD-BCR and participate in induction and regulation of B-cell antigen dependent activation. On the other hand, in case of short-term CD-20 loss, B-cell activation is significantly enhanced. This is due to the CD-20 role in down-regulation of several surface markers. Furthermore, we can also underline IL-6’s role in differentiation and proliferation of B-cells. Together with IL-2 and IL-10, IL-6 induces loss of CD-20 in B-cells, emphasizing its role in initial activation of immune cells. Both processes determine increased plasmablasts proliferation, while IL-6 stimulates the process of differentiation in plasmocytes. IL-2 and IL-10 were also found to induce CD126 expression in plasmablasts [25].
TH17 cells represents a specific type of activated naive T cell. The process of differentiation is initiated by TGF-β and IL-21, while IL-6 and IL-1β allow the amplification of the lineage. Finally, IL-23 determines the pathogenetic role of TH17 cells by stimulating IL-17, IL-21 and IL-22 production. In addition to conventional, antigen-induced Th17 cells—which express TCRs—there are also natural Th17 cells (nTh17), which lack TCRs and thus bypass the classical activation process. This factor denotes the importance of those immune cells in regulation of normal homeostasis. A common finding in human Treg genes is the absence of exon 2 in FoxP3, unbaling Treg to control IL-17+ T-cell proliferation [19, 22-24].
Another important pathogenetic factor is the production of cell-derived microparticles (MPs) by cells exposed to systemic inflammation and oxidative stress. They are released during both cellular activation and cell death, whether by apoptosis or necrosis. An important role in this process plays the Rho-associated kinase 1, which induces the blebbing process of the MP. Besides that, adrenaline, ADP, thrombin, Ca2+, collagen and complement were demonstrated to activate MP formation. It is also believed that their pathogenetic evolution starts with altered lipids distribution in cytolema bilayer. This disbalance is expressed through a low level of cholesterol and phospholipids, with a high expression rate of phosphatidylserine on the outer leaflet. Mostly, they contain cytoplasmic and cytolemic contents of the parent cell, which helps us deduce their origin. The presence of CD4, CD3 or CD8 on the MP surface indicates lymphoid origin while platelet MP are marked by the expression of glycoproteins IIb–IIIa, P-selectin/CD42a/ phosphatidylserine (binding annexin V) [26]. Similarly, endothelial MP displays surface CD31 or CD146. Their size varies between 50-800 nm which makes them detectable by scanning electron microscopy. According to Knijff-Dutmer et al. [27], MP’s concentration in RA is significantly higher, correlating with disease activity. In rheumatoid arthritis, they mainly expose complement components or activator factors. An important point for connecting RA and MP is represented by the presence of citrullinated proteins in MPs, an important pathogenetic factor for rheumatoid arthritis. Platelet derived MP’s have both inflammatory and pro-coagulative effects. On the one hand, secretory PLA2 determines formation of arachidonic acid which is converted in prostaglandins by COX-2, inflammation mediator. On the other hand, PAF is mainly found bound to the PMPs, inducing platelet activation, maintaining local and systemic inflammation/hypercoagulation. Furthermore, P-selectin, a receptor that mediates adhesion of platelets and endothelial cells to the monocytes and granulocytes, is induced by activation of platelets and is found on the surface of PMPs [28]. Additionally, MP interact with factors Va, VIII and IXa, thereby facilitating assembly of prothrombinase complex.
Microparticles role in pathogenesis of vasculitis can be supported by the fact that between 8% and 26% of the endothelial cells internalized MPs and MPs-ICs; a smaller proportion of these cells kept these vesicular structures on their surface (2% to 5%). Both internalization and membrane binding primarily involve the same set of receptors: ICAM-1, CD36 (a scavenger receptor), and CD93 (the C1q receptor). Microparticles complexed with CICs were observed to induce a stronger reaction compared to isolated MPs. This includes increased expression of ICAM-1/2 and supernatant accumulation of IL-6/8 was captured. Besides that, CCL-2/5, a group of chemokines responsible for recruiting and activating immune cells (especially monocytes) in peripheral tissues, were found to be hyper-expressed in endotheliocytes. In addition to the adhesion process to the endothelium being stimulated, intercellular connections between EC are destroyed. This is evidenced by reduced membrane expression of VE-cadherin and depolymerization of actin filaments, which together alter the structure, organization, and continuity of the endothelium in RA. Moreover, MPs are able to activate apoptosis of endothelial cells through unknown mechanisms. The fact that these manifestations are dose-dependent, makes them an important asset for clinicians in future [28].
During systemic inflammation, there are modifications in blood rheology, which also affect vascular endothelial function. Because of increased concentrations of platelets and plasma proteins, along with plasma loss volume, blood becomes more viscous. Additionally, disturbances in vascular smooth muscle tone in addition to increased roughness of endothelium, induce turbulent blood flow. It is believed that endothelial cells possess specialized mechanoreceptors—including components of the cytoskeleton, G-protein coupled receptors (GPCRs), junctional proteins, integrins, and ion channels—that can detect mechanical stimuli such as flow turbulence. Thus, stimulation of NF-kB pathway (c-JUN/AP-1) will determine an increase in adhesives (VCAM-1/E-selectin/ICAM-1) expression. Contrary to pro-inflammatory effects, TNF-α, along with IL-1b induces KLF2 (Kruppel-like factor 2) expression, normally stimulated by laminar flow shear stress. It inhibits the activation of p65 (transcription factor), downregulating VCAM1 and E-selectin transcription. It also upregulates thrombomodulin and NOS3 expression and inhibits transcription of VEGF receptor 2 [13].
Discussion
Due to multiple discoveries in RA and SRV pathogenicity and disease course, we can deduce several pathways leading to vascular damages in this type of vasculitis. All the effects described in this article could be applied both to the healthy and already damaged vessels. Based on RA pathogenicity, all the morphological changes develop at the same time, with rate of progression influenced by genetic predisposition, which causes certain processes to become exacerbated more rapidly than others. This is supported by higher incidence of SRV in RA patients who presented HLA-DRB1*04/04 or HLA-C*03 allele or p53 mutations. In the incipient forms, there is only local evidence of vessel modifications. Vascular endothelium is highly damaged at the site of affected articulations (PIP and MCP). It is favoured by intense inflammatory reactions of Fibroblast-Like Synoviocytes (FLS), ICs formation and deposition, as well as microparticle synthesis. FLS are known to secrete growth factors (TGF-β/FGF), adhesion molecules (VCAM-1, ICAM-1 and integrins) and cytokines (IL-6 and TNF-α) [29]. Entering the systemic circulation, they stimulate their targets, one of them being the endothelial cell of the vascular wall. At this point, IL-6, IL-10 and IL-17 have additive effects. It was observed that all three inflammatory mediators have stimulated ICAM-1 expression, E-selectin was increased in IL-17 and TNF-α stimulation, VCAM-1 was associated with TNF-α. Both IL-6 and IL-17 enhanced cell adhesion through expression of other stimulating Interleukins and molecules. IL-1/CXCL1 and vWF are some of the effectors of the IL-17 mediated adhesion. In addition to that, IL-6 induces IL-8/CXCL8, MCP-1 and CCL2 transcription. Besides interleukins, these effects were observed in the MPs dependent cell activation, where increased ICAM-1/2, P-selectin and CCL-2/5 were observed. All these effects determine immune cells rolling, coupling and transmigration at the site of vascular endothelial cells. Further changes are induced by increased intercellular space between endotheliocytes. It is thought to be related with activation of p38MAPK/ERK pathway through TNFR1- TNF-α complex, which determines rearrangement and destabilization of microtubules. Also, the apoptosis induced by IL-17 and MPs, alongside Ca-CaM dependent MLCK inhibition, speeds up this step. MPs also inhibit VE-cadherin expression and enhance actin filament depolymerization. These changes are important for the translocating mechanism of the cells and ICs to the subendothelial space. Further processes can be more described as a destructive step. It is mainly connected to the ICs activity, as they have the ability both to form and precipitate in the subendothelial space. MET-osis, NET-osis, Citrullination and cell death are some of the pathways which can stimulate ICs. Through MET-osis and NET-osis, macrophages and neutrophils expel their intracellular (MMPs/PAD) enzymes in extracellular space. Besides their damaging action on self-cells, they degradation and citrullination of self-peptides, transforming them in Ag. Also, both necrosis and apoptosis induce Microparticles formation. They can also bind Ab as MPs contain citrullinated molecules. What’s more, all the immune cells present at the site of inflammation express FcR on their membrane. All these factors determine ICs migration and synthesis in the subendothelial space. Lastly, coupling with specific receptors, induces stimulation of different cell processes, for example: ITAM activation increases the phagocytosis rate and cytokine transcription. Besides that, Ag-Ab complexes can bind C1q inducing complement cascade, which determines chemotaxis (C3a, C5a), opsonization (C5b) and cytolysis (through membrane attack complex). At this point, a self-sustaining inflammatory loop is established, wherein cytokines, immune cells, and ICs perpetuate each other’s production and activity. A very important principle that we should not forget is that all those factors have reciprocal potentiating mechanisms, thus inhibition of one of those factors drastically decreases the inflammation.
Conclusions
The study provides a comprehensive analysis of the complex interactions between immune and vascular systems in RA and SRV. EC play a central role in regulating inflammation and coagulation, with activation pathways involving cytokines such as TNF-α, IL-6, and IL-17. These cytokines enhance the expression of adhesion molecules (ICAM-1, VCAM-1, E-selectin), stimulate angiogenesis, and alter endothelial barrier integrity, leading to increased vascular permeability and immune cell recruitment. ICs, microparticles, and systemic inflammatory mediators contribute to the progression of vascular damage by amplifying pro-inflammatory cascades and inducing complement activation, phagocytosis, and cellular apoptosis.
In SRV, the interplay of IC deposition, cytokine release, and immune cell activation perpetuates endothelial dysfunction and hypercoagulability. Specific genetic predispositions, including HLA-DRB1 and HLA-C alleles, were identified as exacerbating factors, linking immune dysregulation to vascular pathology. Novel insights into the role of microparticles and immune cell-derived cytokines further emphasize their pathogenic contributions to endothelial disruption and thrombo-inflammatory processes.
This research highlights critical mechanistic pathways underlying vascular damage in RA and SRV, offering potential targets for therapeutic intervention. It underscores the importance of early detection and precise modulation of inflammatory and coagulative pathways to mitigate vascular complications in autoimmune diseases.
Competing interests
None declared.
Authors’ contributions
Study conception and design: ER, AP, LG. Data acquisition: AP, MS, CN, IL. Analysis and interpretation of data: ER, LC and SA. Drafting of the manuscript: AP, ER. Significant manuscript review with significant intellectual involvement: LG, ER, SA, LC. Approval of the „ready for print” version of the manuscript: ER, LG, LC, SA, MS, CN, IL, AP.
Acknowledgements and funding
The study had no external funding.
Provenance and peer review
Not commissioned, externally peer review.
Authors’ ORCID IDs
Eugeniu Russu – https://orcid.org/0000-0001-8957-8471
Liliana Groppa – https://orcid.org/ 0000-0002-3097-6181
Lia Chișlari – https://orcid.org/ 0000-0002-7088-568X
Svetlana Agachi – https://orcid.org/ 0000-0002-2569-7188
Marius Semionov – https://orcid.org/0009-0007-8749-710X
Chiril Nartea – https://orcid.org/0009-0004-6931-2173
Iosif Leanca – https://orcid.org/0009-0001-9335-3360
Artemie Pastuhov – https://orcid.org/0009-0005-5310-8650
References
Kaye O, Beckers CC, Paquet P, Arrese JE, Piérard GE, Malaise MG. The frequency of cutaneous vasculitis is not increased in patients with rheumatoid arthritis treated with methotrexate. J Rheumatol. 1996 Feb;23(2):253-7.
Makol A, Crowson CS, Wetter DA, Sokumbi O, Matteson EL, Warrington KJ. Vasculitis associated with rheumatoid arthritis: a case-control study. Rheumatology (Oxford). 2014 May;53(5):890-9. doi: 10.1093/rheumatology/ket475.
Mertz P, Wollenschlaeger C, Chasset F, Dima A, Arnaud L. Rheumatoid vasculitis in 2023: changes and challenges since the biologics era. Autoimmun Rev. 2023;22(9):103391. https://doi.org/10.1016/j.autrev.2023.103391.
Laskari K, Ahmadi-Simab K, Lamken M, Csernok E, Gross WL, Hellmich B. Are anti-cyclic citrullinated peptide autoantibodies seromarkers for rheumatoid vasculitis in a cohort of patients with systemic vasculitis? Ann Rheum Dis. 2010 Feb;69(2):469-71. doi: 10.1136/ard.2009.110411. Erratum in: Ann Rheum Dis. 2010 May;69(5):812. Laskaria, K [corrected to Laskari, K].
Makol A, Kermani T, Warrington K, Stone JH, Matteson EL. Rheumatoid vasculitis. In: Stone JH, editor. A clinician's pearls and myths in rheumatology [Internet]. 2nd ed. Cham: Springer; 2023 [cited 2024 Nov 23]. Available from: https://doi.org/10.1007/978-3-031-23488-0_2
Bartels CM, Bridges AJ. Rheumatoid vasculitis: vanishing menace or target for new treatments? Curr Rheumatol Rep. 2010 Dec;12(6):414-9. doi: 10.1007/s11926-010-0130-1.
Wang F, Liu J, Fang Y, Wen J, He M, Han Q, Li X. Hypercoagulability in rheumatoid arthritis: a bibliometric analysis and retrospective data mining study. ACS Omega. 2023;8(50):48522-48534 doi: 10.1021/acsomega.3c08460.
Hunt BJ, Jurd KM. Endothelial cell activation. A central pathophysiological process. BMJ. 1998;316(7141):1328. doi: 10.1136/bmj.316.7141.1328.
Pober J, Sessa W. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7(10):803-815. https://doi.org/10.1038/nri2171.
van Hinsbergh VW. Endothelium - role in regulation of coagulation and inflammation. Semin Immunopathol. 2012 Jan;34(1):93-106. doi: 10.1007/s00281-011-0285-5.
Genta MS, Genta RM, Gabay C. Systemic rheumatoid vasculitis: a review. Semin Arthritis Rheum. 2006;36(2):88-98. https://doi.org/10.1016/j.semarthrit.2006.04.006.
Stone JH. Immune complex-mediated small-vessel vasculitis. In: Firestein GS, Budd RC, Gabriel SE, et al., editors. Kelley and Firestein’s Textbook of rheumatology. 10th ed. Philadelphia: Elsevier; 2017. p. 1571-1580. ISBN 9780323316965.
Urschel K, Cicha I. TNF-α in the cardiovascular system: from physiology to therapy. Int J Interferon, Cytokine Mediator Res. 2015;7:9-25. https://doi.org/10.2147/IJICMR.S64894
Narazaki M, Tanaka T, Kishimoto T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert Rev Clin Immunol. 2017;13(6):535-551. https://doi.org/10.1080/1744666X.2017.1295850
Montgomery A, Tam F, Gursche C, Cheneval C, Besler K, Enns W, et al. Overlapping and distinct biological effects of IL-6 classic and trans-signaling in vascular endothelial cells. Am J Physiol Cell Physiol. 2021 Apr 1;320(4):C554-C565. doi: 10.1152/ajpcell.00323.2020.
Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003;24(1):25-29. https://doi.org/10.1016/S1471-4906(02)00013-3.
Hashizume M, Higuchi Y, Uchiyama Y, Mihara M. IL-6 plays an essential role in neutrophilia under inflammation. Cytokine. 2011 Apr;54(1):92-9. doi: 10.1016/j.cyto.2011.01.007.
Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6(3):315-325. https://doi.org/10.1016/S1074-7613(00)80334-9.
von Vietinghoff S, Ley K. Interleukin 17 in vascular inflammation. Cytokine Growth Factor Rev. 2010 Dec;21(6):463-9. doi: 10.1016/j.cytogfr.2010.10.003.
Zhang L, Liu M, Liu W, Hu C, Li H, Deng J, et al. Th17/IL-17 induces endothelial cell senescence via activation of NF-κB/p53/Rb signaling pathway. Lab Invest. 2021 Nov;101(11):1418-1426. doi: 10.1038/s41374-021-00629-y.
Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. 2023;23(1):38-54. https://doi.org/10.1038/s41577-022-00746-9.
Zhu F, Wang Q, Guo C, Wang X, Cao X, Shi Y, et al. IL-17 induces apoptosis of vascular endothelial cells: a potential mechanism for human acute coronary syndrome. Clin Immunol. 2011 Nov;141(2):152-60. doi: 10.1016/j.clim.2011.07.003.
Hot A, Lenief V, Miossec P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann Rheum Dis. 2012;71(5):768-776. doi: 10.1136/annrheumdis-2011-200468.
Robert M, Miossec P. Effects of Interleukin 17 on the cardiovascular system, Autoimmun Rev. 2017;16(9):984-991. https://doi.org/10.1016/j.autrev.2017.07.009.
Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. 2001;97(6):1817-1822. doi: 10.1182/blood.V97.6.1817.
Ardoin SP, Shanahan JC, Pisetsky DS. The role of microparticles in inflammation and thrombosis. Scand J Immunol. 2007;66:159-165. https://doi.org/10.1111/j.1365-3083.2007.01984.x
Knijff-Dutmer EA, Koerts J, Nieuwland R, Kalsbeek-Batenburg EM, van de Laar MA. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheum. 2002 Jun;46(6):1498-503. doi: 10.1002/art.10312.
Atehortúa L, Rojas M, Vásquez G, et al. Endothelial activation and injury by microparticles in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res Ther. 2019;21(1):34. https://doi.org/10.1186/s13075-018-1796-4
Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010 Jan;233(1):233-55. doi: 10.1111/j.0105-2896.2009.00859.x.