Introduction
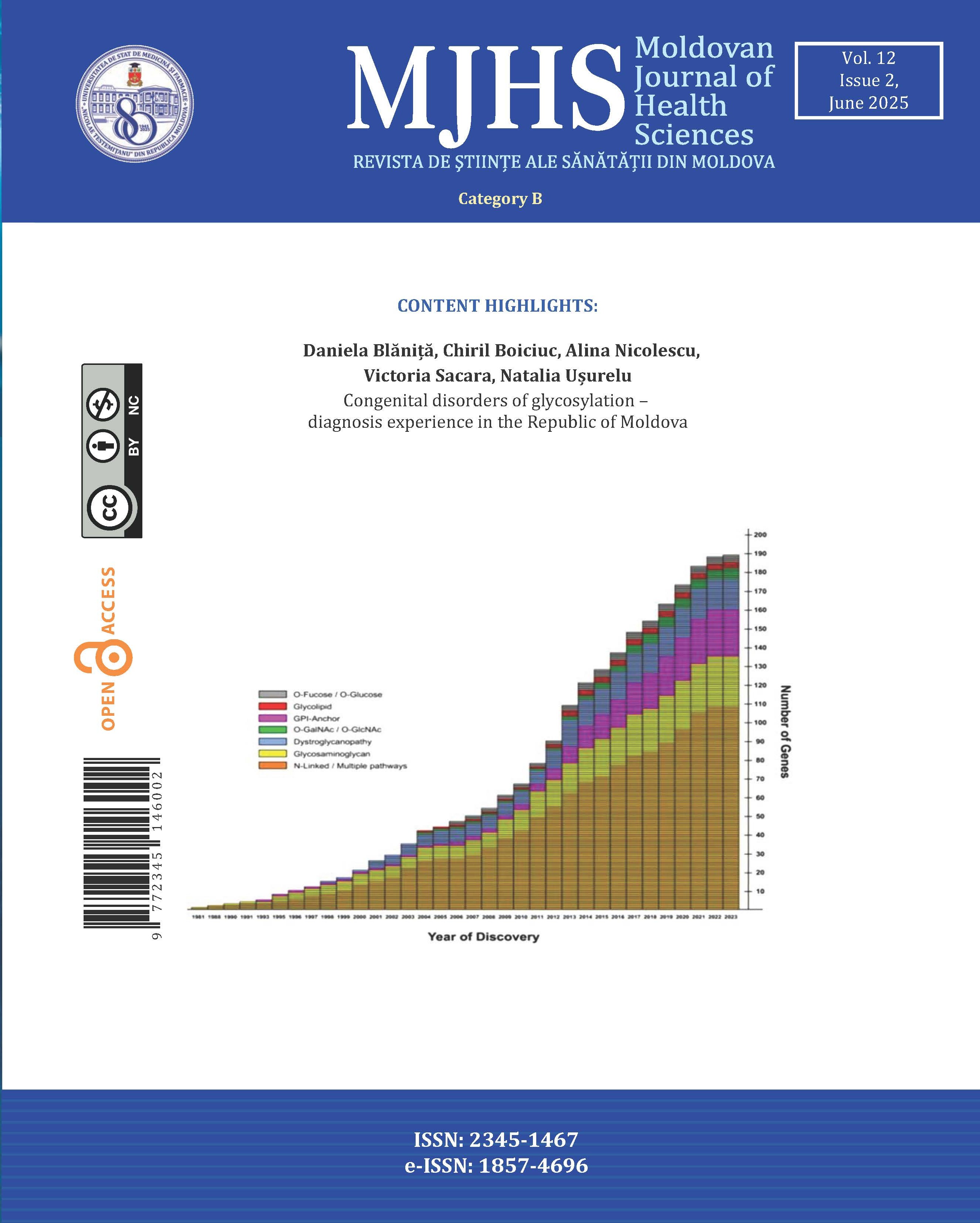
Congenital disorders of glycosylation (CDG) are a group of rare metabolic disorders, with multisystem impairment that result from disturbed protein and lipid glycosylation. The first case was clinically reported in 1980 by Jaeken et al. [1]. With the advent of next-generation sequencing methods in the early 2010s, the identification of CDG types has increased exponentially Currently, there are approximately 200 types of CDG with 189 phenotypes (fig.1) [2, 3]. The prevalence of this group of pathologies is estimated to be less than 1 per 100.000 in Europe [4].
Based on the mode of action, the CDG is classified in 8 categories:
1. Disorders of monosaccharide synthesis and interconversion;
2. Disorders of nucleotide sugar synthesis and transport;
3. Disorders of N-linked protein glycosylation;
4. Disorders of O-linked protein glycosylation;
5. Disorders of lipid glycosylation;
6. Disorders of vesicular trafficking;
7. Disorders of multiple glycosylation pathways;
8. Disorders of glycoprotein/glycan degradation [2].
Considering that CDG is a group of pathologies with multisystem damage, 19 involved organ systems has been described by BG Ng et al. The most affected was the nervous system in 81%, followed by ocular in 46% and muscular in 53%. The dysmorphic features were revealed in 56% [2]. Since 1984, Isoelectric focusing of transferrin (IEFT) has been considered a biochemical method used to detect abnormalities in transferrin glycosylation, making it a key screening tool for diagnosing Congenital Disorders of Glycosylation (CDG), particularly those involving N-glycosylation [1, 5]. IEFT is a specialized electrophoretic technique that separates transferrin isoforms based on their isoelectric point, which depends on the number of sialic acid residues attached to its N-glycans. Following the analysis of IEFT results, two pathological transferrin profiles can be identified: type I and type II. The IEFT type I profile is associated with defects in the endoplasmic reticulum, while the type II profile is linked to abnormalities in the Golgi apparatus. However, normal results do not exclude CDG, as some types may present with mild or absent transferrin abnormalities. This is why IEFT remains an essential first-line biochemical screening tool for CDG but should always be followed by molecular diagnostics for a definitive diagnosis [6].
|
Fig. 1 The exponential character of the identification of CDG types [2]. The number of genes associated with CDG shows a clear upward trend, with a significant increase in reporting starting from 1981 to 2023. The pathways represented by color categories indicate distinct glycosylation types, reflecting the genetic complexity of CDG. O-Fucose/O-Glucose: Pathways involving the attachment of fucose or glucose residues to proteins; Glycolipid: Refers to disorders involving glycosphingolipids or other glycolipid structures; GPI-Anchor: Glycosylphosphatidylinositol anchor defects, where proteins fail to properly attach to cell membranes; O-GalNAc/O-GlcNAc: Pathways associated with O-linked glycosylation involving GalNAc (N-acetylgalactosamine) or GlcNAc (N-acetylglucosamine); Dystroglycanopathy: Disorders affecting glycosylation of dystroglycan, essential for muscle and brain function; Glycosaminoglycan: Pathways related to glycosaminoglycan biosynthesis, affecting connective tissue functions; N-Linked/Multiple Pathways: Defects involving N-linked glycosylation or overlapping multiple glycosylation pathways. |
The objective of this study is to diagnose CDG using Isoelectric Focusing of Transferrin in the Republic of Moldova and to identify diseases that mimic CDG.
Material and methods
The selective cross-sectional observational descriptive study was conducted at the Institute of Mother and Child, in the Laboratory of Prevention of Hereditary Pathologies from October 2018 to April 2024. The inclusion criteria for the study encompassed children with multisystem involvement of unclear etiology, defined by the following characteristics: age between 3 months and 18 years, psychomotor and growth retardation, muscle hypotonia, seizures, dysmorphic features and with chromosomal abnormalities excluded. These manifestations were accompanied by the involvement of other organs and systems, including but not limited to the cardiovascular, hepatic, musculoskeletal, ophthalmologic, dermatologic, immune, and endocrine systems. The exclusion criteria included children with multisystem involvement of confirmed etiology, viral hepatitis, and transferrin polymorphism in IEFT. Following the medical-genetic consultations at the Institute of Mother and Child in the Republic of Moldova and a comprehensive evaluation, including medical history, psychomotor development assessment, physical examination, 320 patients were selected according to the inclusion criteria and enrolled in the study. All patients have signed the consent for participation (approved by Research Ethics Committee of Nicolae Testemițanu State University of Medicine and Pharmacy Minutes No. 45, from July 03, 2019. For each included patient there was completed a questionnaire noting the clinical, phenotypical and investigational features. The biological samples were collected, including serum, plasma, urine, DNA, and DBS. The screening of 150 patients was performed using the IEFT method. For differential diagnosis of other diseases mimicking CDG, metabolic work-up (amino acids in blood, organic acid in urine, acylcarnitine profile) and selective MLPA, CGH-array, and SNP/WES/WGS have been performed. The diagnostic algorithm for CDG and mimicking pathologies was developed in accordance with the innovation act “Algorithm for the evaluation of children with multisystem involvement for the diagnosis of Congenital Disorders of Glycosylation” No. 555 from 29.07.2024. The database has been created using Microsoft Office Excel (Microsoft Corporation, Excel, version 2010, WA, USA), and IBM SPSS Statistics 28.0 program was used for the descriptive statistics to investigate the characteristics of the entire cohort.
Results
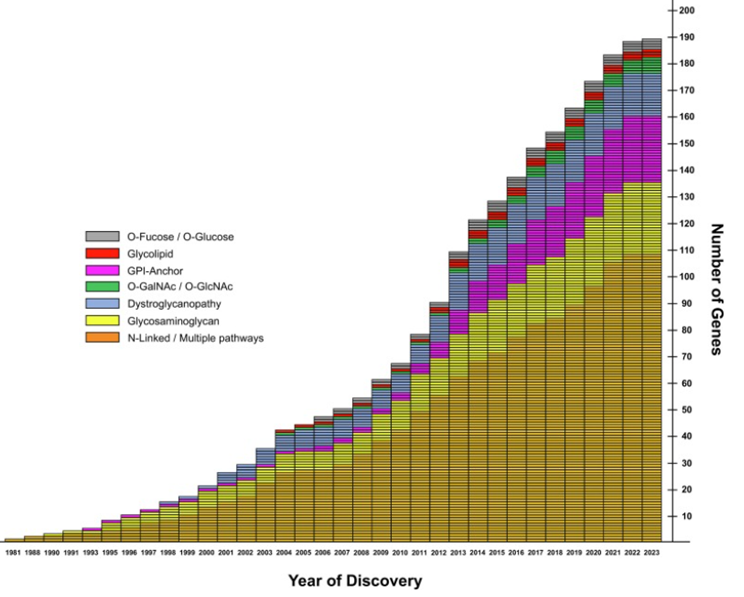
A total of 320 individuals were enrolled in the study. Due to the multisystem involvement, the clinical presentation of the patients included in the research exhibited significant variability. One hundred forty-two individuals (44.4%) were female, and 178 (55.6%) were male. The range of age was around 3-216 months, the mean age was 44 months. The clinical heterogeneity revealed the involvement of 13 affected systems, the leading system being attributed to central nervous system. The second place is hematological system followed by cardiovascular, ophthalmological, gastrointestinal, muscular, skeletal, hepatic, endocrine, renal, urological, auditory, pulmonary and immunological (tab. 1, fig. 2).
|
Fig.2 The multisystem affection in investigated Moldovan cohort suspected for CDG The pie chart illustrates the distribution of multisystem involvement among studied patients. Each segment represents a specific organ or system affected, along with the corresponding percentage of patients exhibiting symptoms in that system. |
The predominant complaints reported by study group showed the following incidences: fatigue in 280 patients (87.5%) (95% CI 0.84-0.91), low attention 250 (78.1%) (95% CI 0.74-0.83), concentration disorders 246 (76.9%) (95% CI 0.73-0.82), verbal retardation 223 (69.7%) (95% CI 0.65 - 0.75), failure to thrive 151 (47.2%) (95% CI 0.43-0.54), dehydration 65 (20.3%) (95% CI 0.17-0.26) and others. Regarding the inclusion criteria in the study, seizures have been registered in 161 patients (50.3%) (95% CI 0.45-0.56), psychomotor retardation – in 275 (85.9%) (95% CI 0.83-0.91), failure to thrive – in 199 (62.2%) (95% CI 0.58-0.68), muscle tone disorders – in 310 (96.9%) (95% CI 0.95-0.99), dysmorphic features – in 197 (9.7%) (95% CI 0.57-0.67), and totally of them 317 (99.1%) (95% CI 0.98-1.00) had multisystem involvement.
After a comprehensive clinical examination, the suspected CDG cases were screened by IEFT. In three cases of 150 patients, positive IEFT has been found. Metabolic work-up and molecular genetic studies were undertaken to finalize the diagnosis. After metabolic work-up, in one case, significant galactose levels in plasma were found, as well as galactitol in urine, suggestive for Galactose troubles. Another two cases revealed no particular metabolites. Then, molecular genetic testing by Sanger sequencing using the Genetic Analyzer 3500, manufacturer Applied Biosystems was undertaken, which revealed mutations in the ALDO B gene in two positive IEFT cases and the GALT gene in the other positive case, validating the diagnosis of Hereditary Fructose Intolerance and Galactosemia, respectively. In other two patients with IEFT negative, a mutation in the GNE gene was discovered by WGS, confirming GNE myopathy (tab.2) [7].
Table 1. Clinical variability in patients enrolled in study. | |||
Affected systems | № patients | Frequency (%) | 95% confidence interval |
Neurological | 295 | 92.2 | 0.18-0.27 |
Hematological | 178 | 55.6 | 0.51-0.62 |
Cardiovascular | 130 | 40.6 | 0.35-0.46 |
Ocular | 124 | 38.8 | 0.34-0.44 |
Gastrointestinal | 94 | 29.4 | 0.24-0.34 |
Muscular | 72 | 22.5 | 0.18-0.27 |
Skeletal | 72 | 22.5 | 0.18-0.28 |
Hepatic | 67 | 20.9 | 0.17-0.26 |
Endocrine | 59 | 18.4 | 0.14-0.23 |
Renal | 35 | 10.9 | 0.07-0.14 |
Genitourinary | 20 | 6.3 | 0.04-0.09 |
Audiological | 19 | 5.9 | 0.03-0.09 |
Respiratory | 12 | 3.8 | 0.02-0.06 |
Immunological | 10 | 3.1 | 0.01-0.05 |
Note: CDG – Congenital disorders of Glycosylations; Nr – number of affections patients; 13 affected systems, the central nervous system being most frequently affected. The least affected systems are urological, audiological, respiratory and immunological. However, their presence still highlights the variability of CDG presentations and the need for a multidisciplinary diagnostic approach. Overall, the table underscores the complexity of CDG, emphasizing the importance of comprehensive clinical evaluations for accurate diagnosis and management. | |||
For 50 cases with negative IEFT, complementary tests such as MLPA (5 cases) and WGS (49 cases) allowed for exclusion of CDG and the confirmatory diagnosis of other diseases. For the remaining cases, it was not possible to obtain additional diagnostic information due to limitations in access to confirmatory testing in our country.
Discussions
CDG constitute a group of multisystem hereditary pathological conditions characterized by dysfunctions in the glycosylation process in the Endoplasmic Reticulum (ER) and the cellular Golgi Apparatus (AG). These dysfunctions affect the biosynthesis of glycoproteins and other glycoconjugates, significantly influencing their functionality. The scientific progress in genetics have facilitated the widespread use of high throughput sequencing methods, allowing scientists to identify the molecular etiologies of CDG and increasing the frequency of diagnosis since the first identifications in the 1980s [8]. Despite advances in the field, the incidence and prevalence of all types of CDG have not yet been precisely established.
Table 2. Types of CDG diagnosed in Republic of Moldova. | ||||
Patients | IEFT results | Gene | Mutations | Disease (ORPHA code) |
P1 | Positive | ALDO B | [c.[113-1-115d/l]/[c.[113-1-115d/l] | Hereditary fructose intolerance (ORPHA:469) |
P2 | Positive | GALT | P.E203L/E203L | Galactosemia (ORPHA:352) |
P3 | Positive | ALDO B | c.[113-1-115d/l]/[524C>A] | Hereditary fructose intolerance (ORPHA:469) |
P4 | Negative | GNE | c.*1014_*1037dupCACACACACACACACACACACACA*/c.1767A>Gp.(=) |
GNE myopathy (ORPHA:602) |
P5 | Negative | GNE | c.173 C>T/c.196G>A | GNE myopathy (ORPHA:602) |
Note: CDG – Congenital disorders of Glycosylations; IEFT – Isoelectric Focusing of transferrin; ORPHA - reference portal for information on rare diseases and orphan drugs; P - number of patients, ALDOB - Aldolase B, Fructose-Bisphosphate, GALT - Galactose-1-Phosphate Uridylyltransferase, GNE - Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase. The total 5 cases of CDG were diagnosed. Three cases were IEFT positive, and two cases IEFT negative. The mutation in ALDO B, GALT and GNEs gene were determined. | ||||
Research in this field has led to conflicting opinions among scientific groups. Until recently, Hereditary Fructose Intolerance and Galactosemia were considered secondary abnormalities of the glycosylation process, being excluded from the list of CDG [9, 10]. However, in May 2024, these conditions were reclassified as CDG spectrum disorders, being attributed to derangements in monosaccharide synthesis and interconversion [2].
In the reported types of CDG, the most prevalent symptoms were neurological (80,5%), followed by dysmorphisms (55.6%), endocrine symptoms (55%), skeletal abnormalities (52.7%), ocular problems (46.3%), digestive issue (34.1%), cardiovascular abnormalities (22.0%), muscular problems (11.7%), respiratory issues (10.2%), genitourinary problems (9.8%), psychiatric symptoms (9.8%), renal complications (8.3%), hair-related issues (6.8%) and dental problems (4.4%) [2, 11].
In Moldovan cohort of CDG suspected patients, the most commonly affected systems were neurological and hematological, followed by cardiovascular, ophthalmological, gastrointestinal, skeletal, hepatic, endocrine, renal, urological, auditory, pulmonary, and immunological. The dental, hair-related, psychiatric, and dermatological symptoms were not reported in our cohort.
Considering that CDG is a group of ultra-rare pathologies with multisystem disorder, often mimicking other genetic diseases, they are frequently underdiagnosed. In the diagnostic process, it is necessary to follow a staged diagnostic algorithm that requires a differential diagnosis to be carried out as meticulously as possible. The most important groups of pathologies that must be excluded are Mitochondrial Diseases and Disorders of Phospholipid Synthesis, both of which also involve multisystem damage [12]. At the same time, dysmorphic features related to a frequency of 55.6% in CDG, can often be found in other pathologies. The presence of fat pads can most commonly be seen in PMM2-CDG, and it has also been reported in Wiedemann-Rautenstrauch syndrome (neonatal progeria). Also, the presence of inverted nipples can occasionally be determined in other genetic diseases than CDG, such as Turner, Smith-Lemli-Opitz, Weaver and Robinow syndrome. Almond-shaped eyes can often be found in Prader-Willi syndrome, which is accompanied by hypotonia and feeding problems in childhood, which can also be seen in CDG [12]. Among the forms of CDG that involve liver damage, the differential diagnosis algorithm includes other various genetic diseases and inborn errors of metabolism as Alpers progressive infantile poliodystrophy, alpha-1 antitrypsin deficiency, cystic fibrosis, Gaucher disease, type IV glycogen storage disorder, glycerol dehydrogenase-1-deficiency-3-phosphate, hemochromatosis, 3-hydroxyacyl-CoA dehydrogenase deficiency, lysosomal acid lipase deficiency, Niemann-Pick disease type A, B, C, peroxisomal diseases, S-adenosine homocysteine hydroxylase deficiency, tyrosinemia type I, and Wilson's disease [13]. Protein-lipid enteropathy, described especially in MPI-CDG, can also be found in severe celiac disease, congenital intestinal diseases and in various genetic diseases (Noon disease, Turner disease, skeletal dysplasia FGFR3). When an endocrine disorder is observed with predominance of hypoglycemia, it is necessary to carry out the differential diagnosis between fatty acid beta-oxidation defects, glycogen storage disorders, the average form of Beckwith-Wiedemann syndrome, congenital hypopituitarism and congenital adrenal hyperplasia [13]. In the presence of renal damage, it is recommended to exclude tubulopathies of metabolic cause, diseases with energy deficit and metabolic disorders of the storage type of complex molecules, as well as other genetic syndromes accompanied by polycystic kidney disease and nephrotic syndrome [12].
One of the objectives of our study was to identify pathologies that mimic the clinical manifestations of CDG. In the analyzed cohort, mitochondrial diseases were diagnosed in 6 cases, representing a significant percentage of the pathologies confused with CDG because of their multisystem affection. Prader-Willi syndrome was identified in 2 cases, and Angelman syndrome, Zellweger syndrome, and Wilson disease were each diagnosed in one case. Other neurological diseases that mimic CDG were determined by next generation sequencing (tab. 3). These genetic pathologies although have clinical manifestations similar to CDG, show distinct pathogenetic mechanisms, emphasizing the importance of a rigorous and differential diagnostic evaluation in the management of these complex conditions.
Table 3. The diseases determined in our cohort that mimic CDG. | |||
Gene | Diseases that mimic CDG | OMIM/ORPHA CODE | Cases |
PPP2R5D | Jordan Syndrome | OMIM 616355/OPRHA 457279 | 1 |
ALDH7A1 | Pyridoxine-dependent epilepsy | OMIM 1617290 /OPRHA3006 | 1 |
TSEN54 | Pontocerebellar hyperplasia type 2A | OMIM 277470/OPRHA 2524 | 2 |
SOX11 | Coffin-Sirris Syndrome | OMIM 615866/ORPHA 1465 | 1 |
AR | Kenedy spinal and bulbar muscular atrophy | OMIM 313200/ORPHA 481 | 1 |
FOXG1 | Congenital Rett Syndrome | OMIM 613454/ORPHA | 1 |
PEX1 | Zelweger Syndrome | OMIM 214100/ORPHA 912 | 1 |
WHSC1 | Wolf-Hirschhorn Syndrome | OMIM 194190/ORPHA 280 | 1 |
ELN | Wiliams Syndrome | OMIM 194050/ORPHA 904 | 1 |
LAMA2 | Congenital muscle wasting with myosin deficiency | OMIM 607855/1ORPHA 258 | 1 |
OPA1 | Mitochondrial DNA depletion syndrome | OMIM 616896/ORPHA 369897 | 1 |
MTATP6 | Sindromul Leigh | OMIM 256000/2ORPHA 506 | 2 |
SCN2A | Epileptic and developmental encephalopathy type 11 | OMIM 613721/ORPHA | 2 |
ANO5 | Autosomal recessive muscular dystrophy of the limbs | OMIM 611307 | 1 |
SMARCAL1 | Schimke Syndrome | OMIM 242900/ORPHA 1830 | 1 |
Deletion of the 15q11 region | Angelman Syndrome | OMIM 105830/ ORPHA 72 | 1 |
Deletion 15q11-q13 | Prader-Willi syndrome | OMIM 176270/ORPHA 739 | 2 |
GBA | Gaucher disease type 2 | OMIM 230900 /ORPHA 77260 | 1 |
SMN1 | Spinal muscular atrophy | OMIM 253300/ORPHA 253300 | 2 |
RALA | Hiatt-Neu-Cooper syndrome | OMIM 619311/ORPHA 528084 | 1 |
PAH | Phenylketonuria | OMIM 261600/ORPHA 79254 | 3 |
ATP7B | Wilson disease | OMIM 277900/ORPHA 509 | 1 |
SCN8A | Cognitive impairment with or without cerebellar ataxia | OMIM 614306 | 2 |
HFE | Hemochromatosis | OMIM 465508/ORPHA235200 | 1 |
TWNK | Progressive external ophthalmoplegia with mitochondrial DNA deletions | OMIM 609286/ORPHA254892 | 1 |
SCNA1 | Epileptic encephalopathy, type 6 | OMIM619317/ORPHA36387 |
|
NSD1 | SOTOS syndrome | OMIM617169/ORPHA 821 | 1 |
TGFBR1 | LOEYS-DIETZ syndrome | OMIM613795 /ORPHA60030 | 1 |
mtTL1 | MELAS disease | OMIM 540000 /ORPHA 550 | 1 |
SLC9A3R1 | Nephrolithiasis/Hypophosphatemic osteoporosis type 2 | OMIM612286/ORPHA 244305 | 1 |
Note: OMIM - Online Mendelian Inheritance in Man is a knowledgebase of human genes and genetic disorder; ORPHA - reference portal for information on rare diseases and orphan drugs; CDG – Congenital disorders of glycosylations. The table presents the diagnosed pathologies that mimicked CDG, highlighting the mutations identified in the genes associated with each condition, as well as the number of observed cases. | |||
According to reported data, IEFT identifies alterations in the transferrin profile in only 60% of cases, primarily in the presence of N-glycosylation defects, however, a normal IEFT result does not rule out CDG [14]. Therefore, in cases with strong clinical and biochemical suspicion, advanced diagnostic approaches such as next-generation sequencing (NGS), including targeted gene panels or whole-exome/genome sequencing (WES/WGS), are recommended as a definitive diagnostic strategy [14, 15]. The remaining 265 cases did not reach a definitive diagnosis due to limited access to diagnostic methods in the Republic of Moldova, as these are very expensive and not performed in our country. In this context, we cannot exclude the possibility that other types of CDG, which test negative on IEFT, may be present in these 265 cases.
Conclusions
Our study revealed significant clinical diversity among patients with suspected CDG, with multisystem involvement and a predominance of central nervous system involvement. Despite the high frequency of symptoms associated with CDG, only three cases were confirmed by IEFT and genetic testing, with final diagnoses of Galactosemia and Hereditary Fructose Intolerance, representing the first group of glycosylation impairment in the novel classification. In addition, two cases were diagnosed with GNE myopathy. Among 50 cases with negative IEFT result, complementary molecular tests allowed the exclusion of CDG and the establishment of other diagnoses. This emphasizes the complexity of diagnosing CDG and the need for a rigorous diagnostic protocol, including advanced metabolic and genetic testing.
Competing interests
None declared.
Authors’ contribution
DB conceived conceptualization, methodology, data collection, analysis and interpretation, writing – original draft preparation. CB analyzed the result of screening by IEFT. AN – supervision on differential diagnosis data. VS – coordinator of genetic analysis. NU – research coordinator, conceived writing review and editing, validation. The authors read and approved the final version of the manuscript.
Patient consent
Obtained
Ethics approval
This study was approved by the Research Ethics Committee of Nicolae Testemițanu State University of Medicine and Pharmacy (Act No. 45, from July 03, 2019).
Acknowledgments and funding
The research was initiated by the scientific project 18.80012.04.04F, 2018-2019 (CDGSCREEN) and followed by the project 20.80009.8007.22, 07-PS, 2020-2023 (SCREENGEN) financed by the National Agency of Research and Development and Ministry of Education and Research of Republic of Moldova. The research was supported by the grant from the Ministry of Research, Innovation and Digitalization, CNCS-UEFISCDI, project PN-IV-P8-8.3-ROMD-2023-0249 (DiMoMeD), within PNCDI IV (Romania).
Provenance and peer review
Not commissioned, externally peer review.
Authors’ ORCID IDs
Daniela Blăniță – https://orcid.org/0000-0001-7736-3406
Chiril Boiciuc – https://orcid.org/0000-0002-7273-2492
Victoria Sacara – https://orcid.org/0000-0001-9200-0494
Alina Nicolescu – https://orcid.org/ 0000-0001-7022-8893
Natalia Ușurelu – https://orcid.org/0000-0001-8685-3933
References
Jaeken J, Vanderschueren-Lodeweyckx M, Casaer P, Snoeck L, Corbeel L, Eggermont E, et al. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG-deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome? Pediatr Res. 1980;14(2):179. doi: 10.1203/00006450-198002000-00117.
Ng BG, Freeze HH, Himmelreich N, Blau N, Ferreira CR. Clinical and biochemical footprints of congenital disorders of glycosylation: proposed nosology. Mol Genet Metab. 2024 May;142(1):108476. doi: 10.1016/j.ymgme.2024.108476.
Lam C, Scaglia F, Berry GT, Larson A, Sarafoglou K, Andersson HC, et al. Frontiers in congenital disorders of glycosylation consortium, a cross-sectional study report at year 5 of 280 individuals in the natural history cohort. Mol Genet Metab. 2024 Aug;142(4):108509. doi: 10.1016/j.ymgme.2024.108509.
Granjo P, Pascoal C, Gallego D, Francisco R, Jaeken J, Moors T, et al. Mapping the diagnostic odyssey of congenital disorders of glycosylation (CDG): insights from the community. Orphanet J Rare Dis. 2024 Nov 1;19(1):407. doi: 10.1186/s13023-024-03389-2.
Blanita D, Boiciuc C, Turcan D, Sacara V, Usurelu, N. The screening by isoelectric focusing of transferrin for the diagnosis of congenital disorders of glycosylation. Mold Med J. 2021;64(4):50-54. https://doi.org/10.52418/moldovan-med-j.64-4.21.09.
Magalhães APPS, Burin MG, Souza CFM, de Bitencourt FH, Sebastião FM, Silva TO, et al. Transferrin isoelectric focusing for the investigation of congenital disorders of glycosylation: analysis of a ten-year experience in a Brazilian center. J Pediatr (Rio J). 2020;96(6):710-716. https://doi.org/10.1016/j.jped.2019.05.008.
Blăniță D, Boiciuc C, Samohvalov E, Sacara V, Barbova N, Hadjiu S, et al. Challenges in clinical consideration for congenital disorders of glycosylation. Bull Perinatol (Chisinau). 2020;(1):18-22.
Jaeken J, Carchon H. The carbohydrate-deficient glycoprotein syndromes: an overview. J Inherit Metab Dis. 1993;16(5):813-20. doi: 10.1007/BF00714272.
Quintana E, Sturiale L, Montero R, Andrade F, Fernandez C, Couce ML, et al. Secondary disorders of glycosylation in inborn errors of fructose metabolism. J Inherit Metab Dis. 2009 Dec;32 Suppl 1:S273-8. doi: 10.1007/s10545-009-1219-4.
Maratha A, Colhoun HO, Knerr I, Coss KP, Doran P, Treacy EP. Classical galactosaemia and CDG, the N-glycosylation interface. A review. JIMD Rep. 2017;34:33-42. doi: 10.1007/8904_2016_5.
Miller BS, Freeze HH. New disorders in carbohydrate metabolism: congenital disorders of glycosylation and their impact on the endocrine system. Rev Endocr Metab Disord. 2003;4(1):103-113. https://doi.org/10.1023/A:1021883605280.
Altassan R, Péanne R, Jaeken J, Barone R, Bidet M, Borgel D, et al. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: diagnosis, treatment and follow up. J Inherit Metab Dis. 2019;42(1):5-28. doi: 10.1002/jimd.12024.
Cechova A, Altassan R, Borgel D, Bruneel A, Correia J, Girard M, et al. Consensus guideline for the diagnosis and management of mannose phosphate isomerase-congenital disorder of glycosylation. J Inherit Metab Dis. 2020;43(4):671-693. doi: 10.1002/jimd.12241.
Francisco R, Marques-da-Silva D, Brasil R, Pascoal C, dos Reis Ferreira V, Morava E, et al. The challenge of CDG diagnosis. Mol Genet Metab. 2019;126(1):1-5. doi: 10.1016/j.ymgme.2018.11.003.
Blăniță D, Boiciuc C, Stamati A, Hadjiu S, Țurea V, Morava E, Ușurelu N. Screening-ul IEFT în diagnosticul tulburărilor congenitale ale proceselor de glicozilare [IEFT screening in the diagnosis of congenital disorders of glycosylate processes]. In: National Conference “Rare Disease Day 2023”, 2023 Feb 28; Chişinău. Chişinău; 2023. p. 12-16. Romanian.