
Introduction
Male infertility refers to a man's inability to achieve pregnancy within a couple, despite the presence of a fertile female partner. It has been well established that male fertility is primarily dependent on both the quantity and quality of semen. When the volume of ejaculated semen is reduced or of poor quality, the likelihood of natural conception is significantly diminished. According to the World Health Organization (WHO), male-factor infertility is defined as an abnormality in sperm concentration, motility, and/or morphology in at least one of two semen samples, collected at intervals of one to four weeks. Therefore, men whose semen parameters fall below the WHO-established reference values are considered to have impaired fertility [1-3].
The most severe form of male infertility is azoospermia, which refers to the complete absence of spermatozoa in the collected or ejaculated semen. Azoospermia affects approximately 1% of the general male population, while its prevalence among infertile men ranges from 10% to 15% [4].
Historically, men diagnosed with azoospermia were classified as having irreversible infertility, and the use of donor sperm was considered one of the most viable reproductive options. However, with the advent of advanced assisted reproductive technologies (ART) such as intracytoplasmic sperm injection (ICSI), testicular sperm extraction (TESE), and microsurgical TESE (micro-TESE), even men with the most severe forms of azoospermia may now achieve biological parenthood [5].
The frequency of chromosomal abnormalities is inversely correlated with sperm concentration. Among patients with moderate oligozoospermia, chromosomal anomalies are detected in approximately 4% of cases [6]. In contrast, among azoospermic patients, the prevalence of chromosomal alterations ranges from 15% to 25%, depending on the specific subgroups studied [7]. Given the high incidence of chromosomal abnormalities among men with non-obstructive azoospermia and moderate oligozoospermia, karyotype analysis is recommended as the first-line genetic test for patients with quantitative spermatogenesis disorders. Furthermore, this genetic test is also indicated in men with a family history of recurrent miscarriage, congenital malformations, cognitive developmental disorders, or infertility, regardless of semen concentration [6].
The identification of an abnormal karyotype is of great importance for comprehensive and effective genetic counseling, which should provide detailed information regarding the type of chromosomal anomaly or polymorphism, its clinical significance, inheritance pattern, genetic risk to offspring, and available options for prenatal diagnosis.
Genetic counseling plays a critical role in guiding infertile couples to make informed decisions regarding assisted reproductive choices. Therefore, cytogenetic screening remains a key component in the accurate diagnosis, evaluation, prognostic assessment, and successful treatment of male infertility. The objective of the study was to evaluate the profile of chromosomal variations in infertile men with azoospermia, in order to optimize assisted reproductive strategies in infertile couples.
Material and methods
The study was conducted on a cohort of 96 male patients diagnosed with azoospermia, who presented for infertility evaluation. The diagnosis of azoospermia was established based on at least two consecutive semen analyses confirming the complete absence of spermatozoa in the ejaculate. Semen samples were collected in accordance with WHO guidelines, following a recommended abstinence period of 3-7 days. The mean age of the men with azoospermia in the entire sample (n = 96) was 33.8 ± 5.3 years (95% CI: 32.7 – 34.9; median: 33.0). The mean duration of infertility across the cohort was 6.5 ± 4.6 years (95% CI: 5.6 – 7.5).
The cytogenetic analysis was performed at the Cytogenetics Laboratory of the Mother and Child Institute, Center for Reproductive Health and Medical Genetics, Republic of Moldova. All patients underwent classical cytogenetic analysis using peripheral blood lymphocytes. Blood samples were cultured and enriched with phytohemagglutinin to stimulate cell division. After 72 hours of incubation, metaphase was arrested with colchicine, followed by hypotonic shock and fixation with methanol-acetic acid. Karyotyping was performed and interpreted according to the 2016 International System for Human Cytogenetic Nomenclature (ISCN). Between 10 and 30 metaphases were analyzed per patient, with 5 to 10 karyotyped, increasing accordingly in the presence of abnormal cells. In cases with atypical findings, such as suspected sex reversal (46,XX with male phenotype), complementary FISH (Fluorescent In Situ Hybridization) analyses were performed for the SRY and DXZ1 (X) and DYZ1 (Y) markers, along with molecular PCR testing for Y chromosome-specific markers (AZFa, AZFb, and AZFc regions).
All patients underwent endocrine evaluation by measuring serum levels of the following hormones: follicle-stimulating hormone (FSH), luteinizing hormone (LH), prolactin, total testosterone, and free testosterone.
Statistical analysis was performed using SPSS (Statistical Package for the Social Sciences) version 22.0. Comparison of mean parameters between patient groups (with sex chromosome vs. autosomal abnormalities) was carried out using the ANOVA (analysis of variance) test. Differences were considered statistically significant at a p-value < 0.05.
Results
Cytogenetic analysis performed on 96 infertile men with azoospermia revealed a normal karyotype 46,XY in 75% of cases (n = 72), while 25% (95% CI: 16.3–33.7; n = 24) exhibited chromosomal numerical or structural abnormalities. The prevalence of sex chromosome abnormalities was identified in 16.7% of cases (n = 16), whereas 8.3% of cases (n = 8) presented autosomal abnormalities (Fig. 1).
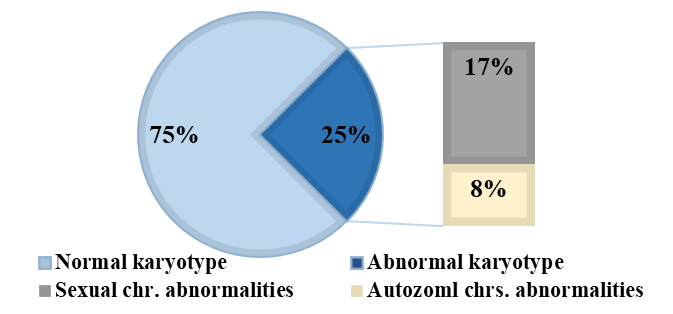 |
Fig. 1 Proportion of chromosomal abnormalities in men with azoospermia (n = 96) Note: The figure illustrates the distribution of chromosomal findings among azoospermic men. A normal karyotype was identified in 75% of cases. Chromosomal abnormalities were found in 25% of the cohort, with 17% involving sex chromosomes and 8% involving autosomal chromosomes. The inner bar chart expands the abnormal karyotype group, distinguishing between sex chromosome abnormalities and autosomal abnormalities. Data are expressed as percentages of the total number of patients. |
A comparative analysis of patient age revealed a statistically significant difference between the two groups. The mean age in the group with sex chromosome abnormalities (n = 16) was 36.2 ± 4.9 years (IQR: 32.5–39.0; median: 35.5), compared to 32.0 ± 2.9 years (IQR: 29.5–34.5; median: 32.0) in the group with autosomal chromosomal abnormalities (n = 8) (F = 4.917; p = 0.037). No statistically significant difference was found in semen volume between the two groups (F = 0.601; p = 0.447). The sex chromosome group had a mean volume of 2.2 ± 0.7 mL (IQR: 1.5–2.5; median: 2.2), while the autosomal group had a mean of 2.4 ± 0.8 mL (IQR: 2.0–2.9; median: 2.4). In contrast, FSH levels differed significantly between groups (F = 7.406; p = 0.012). Patients with sex chromosome abnormalities showed a mean FSH level of 15.4 ± 10.3 IU/L (IQR: 7.5–20.6; median: 15.5), whereas those with autosomal abnormalities had a mean of 4.9 ± 4.8 mIU/mL (IQRR: 1.8–6.7; median: 2.9). Similarly, LH levels were significantly higher in the sex chromosome group (F = 6.576; p = 0.018), with a mean of 15.3 ± 8.9 mIU/mL (IQR: 8.1–23.3; median: 13.0) compared to 6.7 ± 4.0 mIU/mL (IQR: 3.9–9.2; median: 5.8) in the autosomal group. Prolactin levels did not differ significantly between groups (F = 1.158; p = 0.294). The mean prolactin concentration was 12.9 ± 6.5 ng/mL (IQR: 7.0–19.1; median: 11.3) in the sex chromosome group and 15.9 ± 5.6 ng/mL (IQR: 11.3–19.6; median: 18.0) in the autosomal group. Testosterone levels also showed no statistically significant differences (F = 1.224; p = 0.281), with a mean of 3.4 ± 0.9 ng/mL (IQR: 2.9–3.8; median: 3.4) in the sex chromosome group and 3.9 ± 1.2 ng/mL (IQR: 2.8–4.9; median: 3.7) in the autosomal group (Table 1).
Table 1. Characteristics of sex chromosome abnormalities compared to autosomal abnormalities in patients with azoospermia | ||||
Parameters | sex chromosem ab. | Autosomal chrs. ab. | Total | p |
No. of patients | n = 16 | n = 8 | n = 24 |
|
Age (years) | 36.2 ± 4.9 35.5 (32.5-39.0) | 32.0 ± 2.9 32.0 (29.5-34.5) | 34.8 ± 4.8 34.5 (31.0-36.0) | p=0.037
|
Volume (mL) | 2.2 ± 0,7 2.2 (1.5-2.5) | 2.4 ± 0.8 2.4 (2.0-2.9) | 2.2 ± 0.7 2.2 (1.8-2.6) | p=0.447
|
FSH (mIU/mL) | 15.4 ± 10.3 15.5 (7.5-20.6) | 4.9 ± 4.8 2.9 (1.8-6.7) | 11.9 ± 10.1 9.2 (2.9-19.1) | p=0.012
|
LH (mIU/mL) | 15.3 ± 8.9 13.0 (8.1-23.3) | 6.7 ± 4.0 5.8 (3.9-9.2) | 12.4 ± 8.6 9.9 (5.9-19.5) | p=0.018
|
Prolactin (ng/mL) | 12.9 ± 6.5 11.3 (7.0-19.1) | 15.9 ± 5.6 18.0 (11.3-19.6) | 13.9 ± 6.2 11.8 (7.9-19.5) | p=0.294
|
Testosterone (ng/mL) | 3.4 ± 0.9 3.4 (2.9-3.8) | 3.9 ± 1.2 3.7 (2.8-4.9) | 3.6 ± 1.0 3.4 (2.9-4.1) | p=0.281
|
Free testosterone (ng/mL) | 7.1 ± 6.8 2.8 (2.8-8.4) | 15.9 ± 10.5 15.9 (8.4-23.3) | 9.6 ± 8.3 8.4 (2.8-18.5) | p=0.231
|
Note: Chrs. ab.- chromosomal abnormalities; FSH - follicle-stimulating hormone; LH - luteinizing hormone; mL – milliliters, n - number of cases. Statistical analysis: The Student’s t-test was used for comparison of normally distributed continuous variables Mean ± SD (Standard Deviation). The Mann–Whitney U test was applied for non-normally distributed variables Median, IQR (Interquartile Range). A p-value < 0.05 was considered statistically significant. | ||||
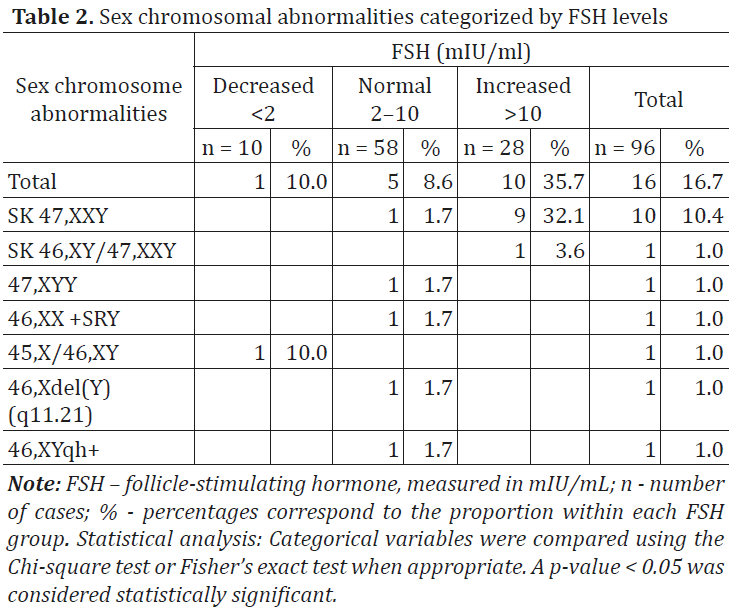
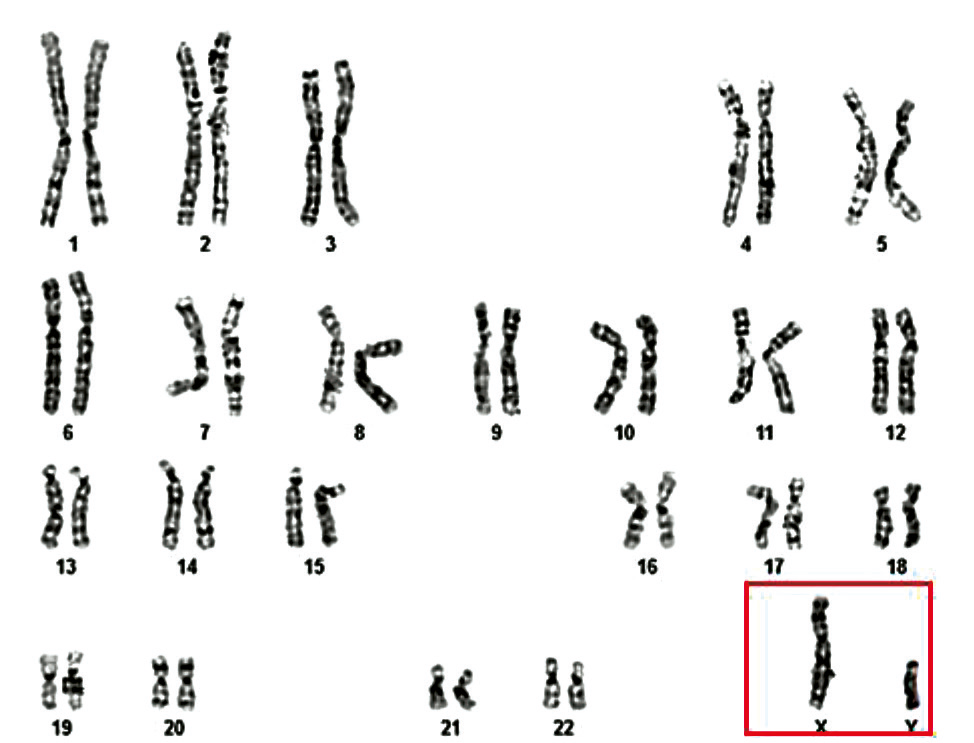
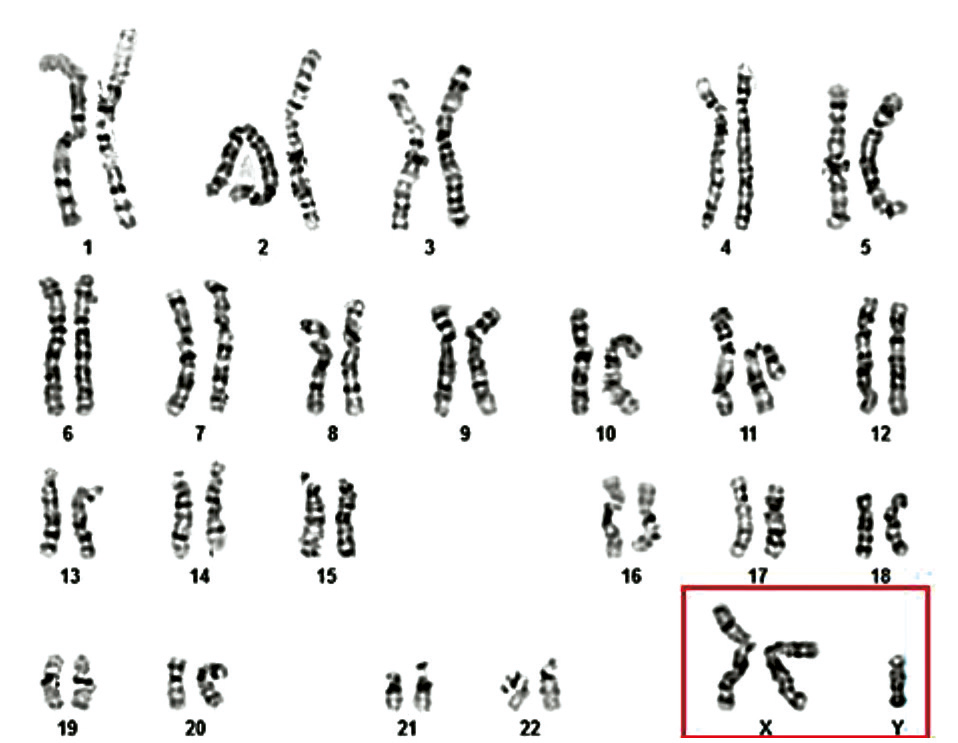
From the total cohort of azoospermic men who underwent cytogenetic investigation (n = 96), 11 patients (11.5%) were diagnosed with X disomy. According to cytogenetic results, the most frequently identified chromosomal variant among these 11 infertile patients with Klinefelter syndrome (KS) was the homogeneous form of trisomy 47,XXY (10 cases – 90.9%), followed by the mosaic form of the same classical variant 47,XXY/46,XY (1 case – 9.1%) (Table 2, Fig. 2 and 3).
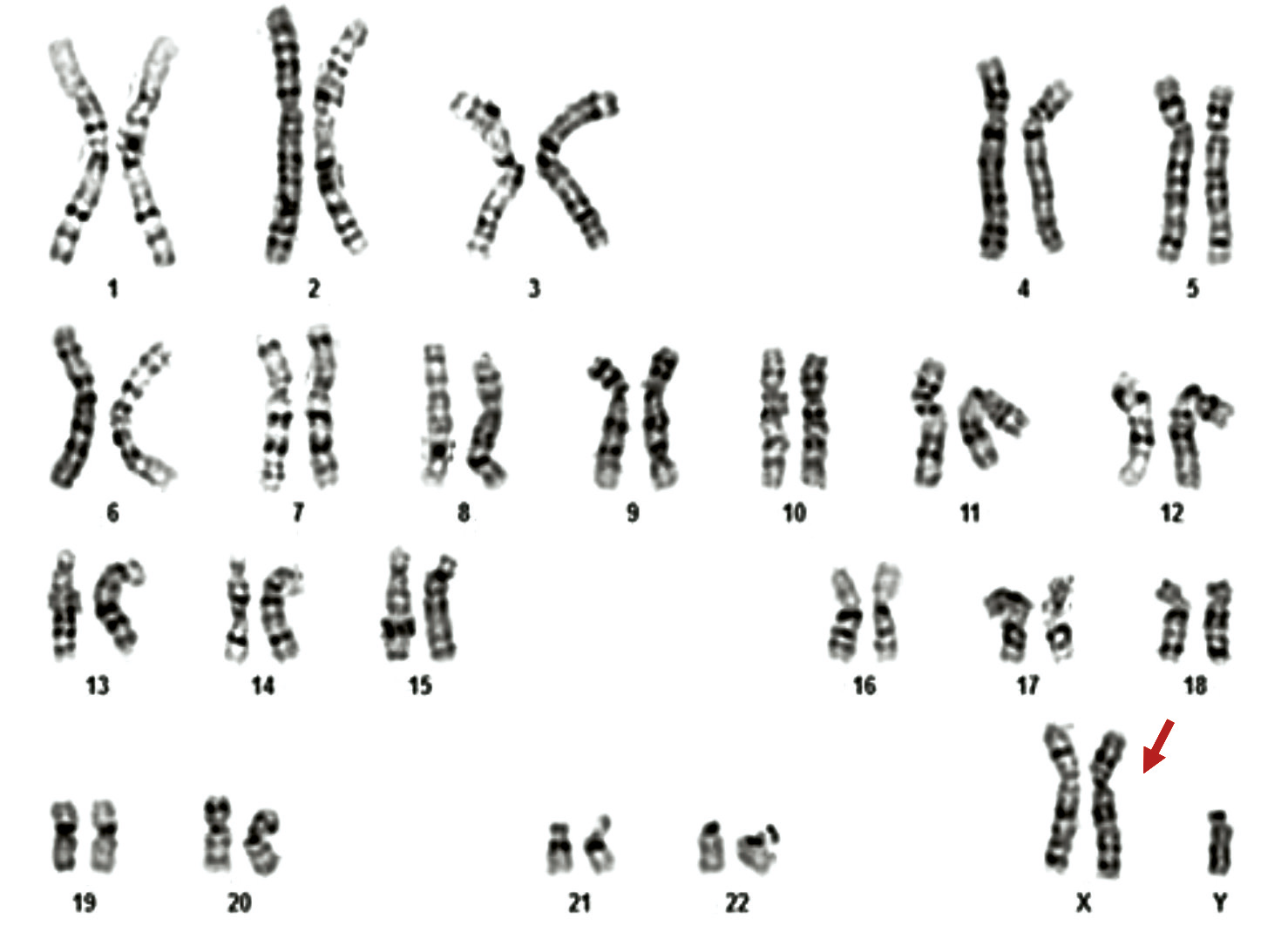 |
Fig. 2 Cytogenetic variant – classical form of Klinefelter syndrome, 47,XXY in a 35-year-old patient Note: The image presents a G-banded karyotype demonstrating the presence of an extra X chromosome, consistent with Klinefelter syndrome. A total of 47 chromosomes are identified, including two X chromosomes and one Y chromosome. The arrow indicates the additional X chromosome. |
A | B |
Fig. 3 Cytogenetic variant – mosaic form of Klinefelter syndrome 46,XY/47,XXY in a 31-year-old patient Note: The figure shows two karyotypes (A and B) illustrating the mosaicism detected in a male patient. Image A displays a normal male karyotype (46,XY), while image B shows a cell line with an additional X chromosome (47,XXY). This cytogenetic mosaicism indicates the coexistence of two distinct cell lines within the same individual. | |
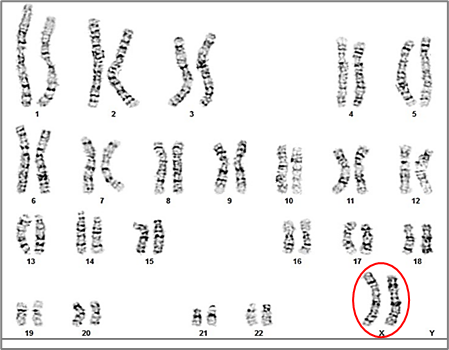
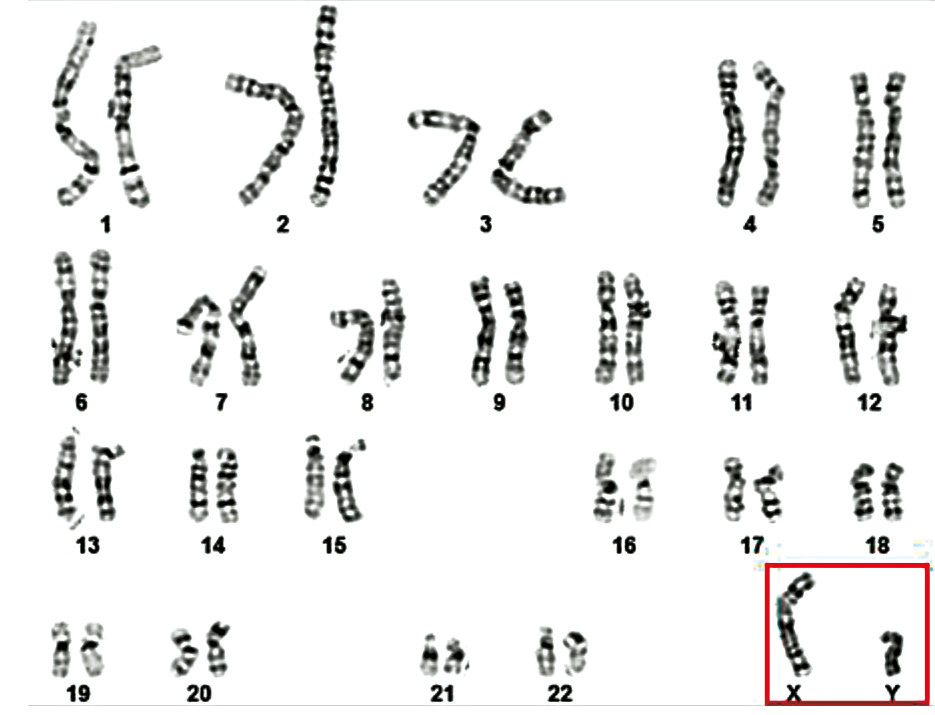
Additionally, one case of each of the following rare sex chromosome abnormalities were identified (1.04% per abnormality): Y disomy (47,XYY), sex reversal (46,XX + SRY), mixed gonadal dysgenesis in a male (45,X/46,XY), and two structural Y chromosome variation, 46,X,del(Y)(q11.21) (Y≤21) and 46,XYqh+ (Y ≥ 18) (Table 2, Fig. 4-7).
|
Fig. 4 Karyotype 47,XYY of a 36-year-old patient [8] Note: The karyotype reveals the presence of an extra Y chromosome, resulting in a 47,XYY karyotype, known as Jacobs syndrome. The red arrow indicates the second Y chromosome. |
The patient presented with a male phenotype, but had a 46,XX karyotype characteristic of a female chromosomal pattern (Fig. 5). A total of 33 metaphases were analyzed, of which 10 were karyotyped, with a banding resolution level of 575-700, which revealed no evidence of mosaicism or other structural or numerical abnormality.
In addition, a FISH (Fluorescence in situ Hybridization) test was performed using probes for the X chromosome (DXZ1, Xp11.1–q11.1) and the Y chromosome (SRY, Yp11.32, and DYZ1, Yq12). The FISH analysis revealed two signals for the X chromosome (DXZ1) and one signal for the SRY gene (Yp11.32).
Molecular analysis of Y chromosome-specific markers – SY81, SY84, sDBY1, and sY620 from the AZFa region; SY127, SY134, sY117, and sY143 from the AZFb region; SY254, SY255, sY153, and sY158 from the AZFc region – showed their absence in the patient's DNA. However, the presence of ZFX and SRY was confirmed. These results support the diagnosis of an XX male with the presence of the SRY gene, but without other Y-specific markers (Fig. 6).
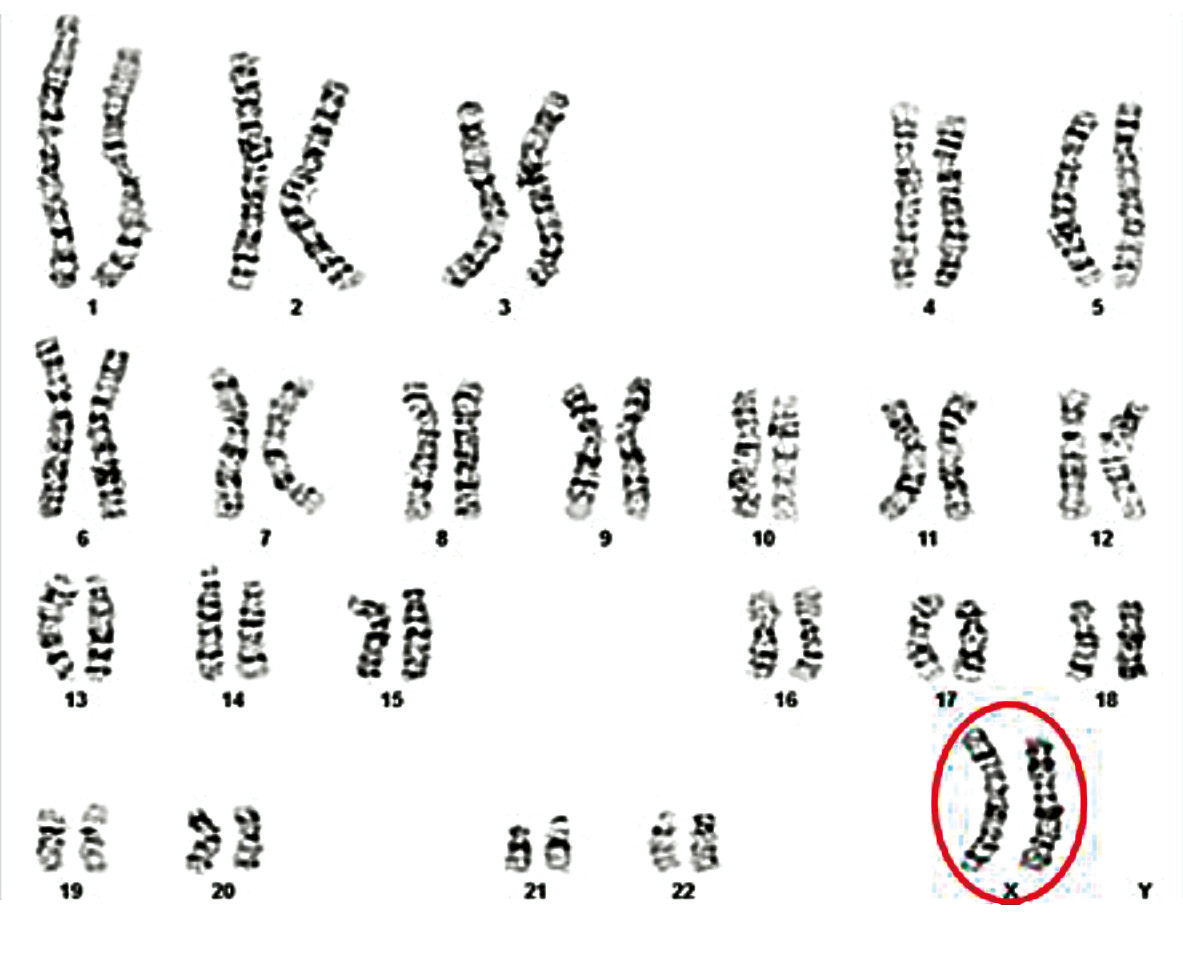 |
Fig. 5Karyotype 46,XX in all cells analyzed in a 31-year-old man [9] Note: The patient, exhibiting a male phenotype, presented with a 46,XX karyotype – comprising 46 chromosomes, including two X chromosomes in the sex chromosome pair – consistent with a female chromosomal pattern in all analyzed cells. (Metaphases counted: 33; Karyotyped metaphases: 10; Band resolution level: 575-700) |
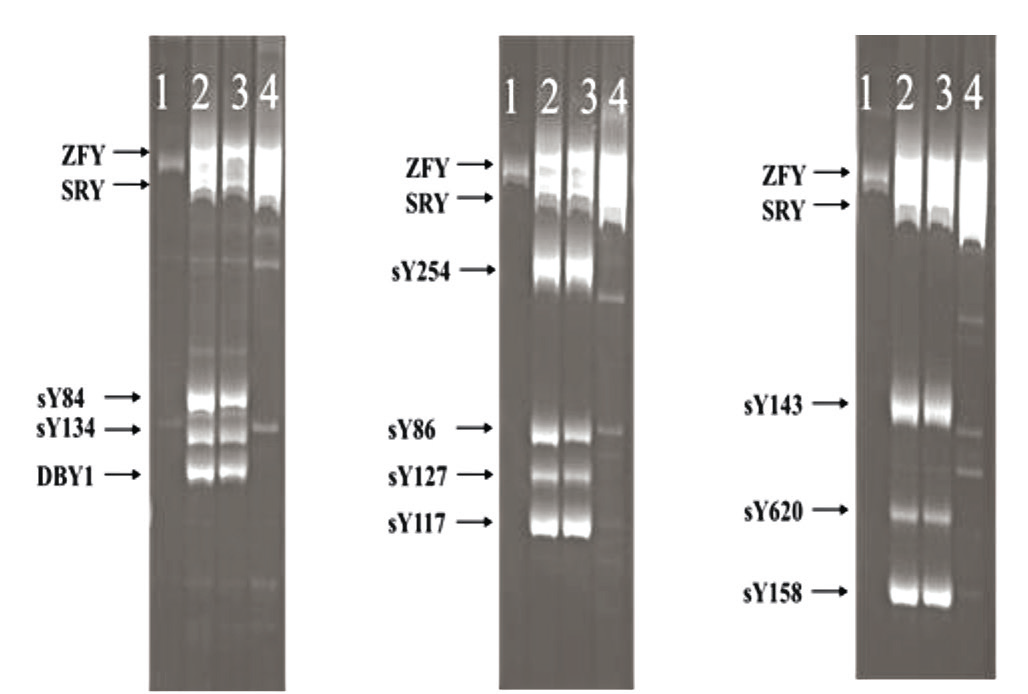 |
Fig. 6 The result of electrophoresis for detection of Y markers in the man with 46,XX [9] Note: 1 – Female control; 2, 3 – Normal male; 4 – Male with deletions of the AZF (a, b, c) region |
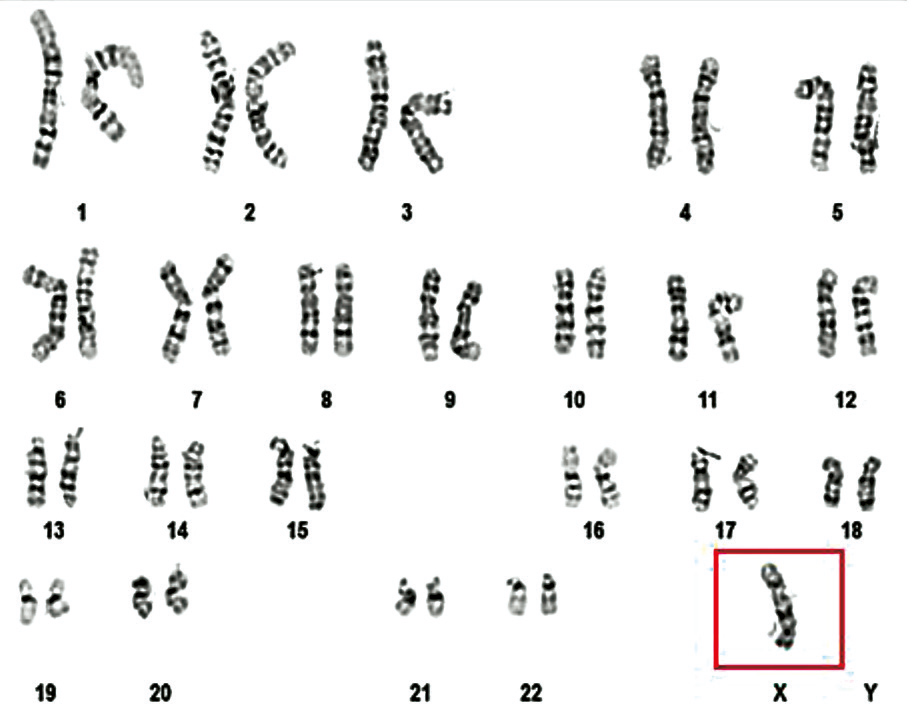
In the patient with a 45,X/46,XY karyotype, two distinct cell lines were identified (chromosomal mosaicism): one cell line with 45 chromosomes, containing a single X chromosome in the sex chromosome pair (X monosomy – 45,X), observed in 3 analyzed cells (20%); and an apparently normal male cell line with 46 chromosomes (46,XY), observed in 12 analyzed cells (80%). The cytogenetic result, according to the 2016 International System for Human Cytogenetic Nomenclature, was 45,X/46,XY (Fig. 7).
A | B
|
Fig. 7 Mosaic karyotype 45,X(3)/46,XY(12) of a 46-year-old patient [10] Note: The mosaic karyotype have two different cell lines: A: 45,X, characterized by monosomy X, which is commonly associated with features of Turner syndrome, was observed in 3 analyzed cells (20%). B: 46,XY, representing a normal male karyotype, cell line was observed in 12 cells (80%). The presence of this mosaicism can result in a wide spectrum of clinical presentations, depending on the distribution and proportion of each cell line in various tissues. In this case, the patient presents an apparently normal male phenotype, likely due to the predominance of the 46,XY cell line, which accounts for 80% of the analyzed cells. | |
Cytogenetic testing of azoospermic patients identified structural variations in autosomal chromosomes in 8.3% of cases (n = 8). These included a variety of chromosomal rearrangements such as translocations 46,XY,t(1;19)(23.2q:q12.4); 46,XY,der(5),t(9;5); 45,XY,rob(13/14); 46,XY,inv(9)(p11q12); 46,XY,inv(9)(p13.21); 46,XY,15ps+; 46,XY,22sts; 46,XY,fra(17)(p12) (Table 3).
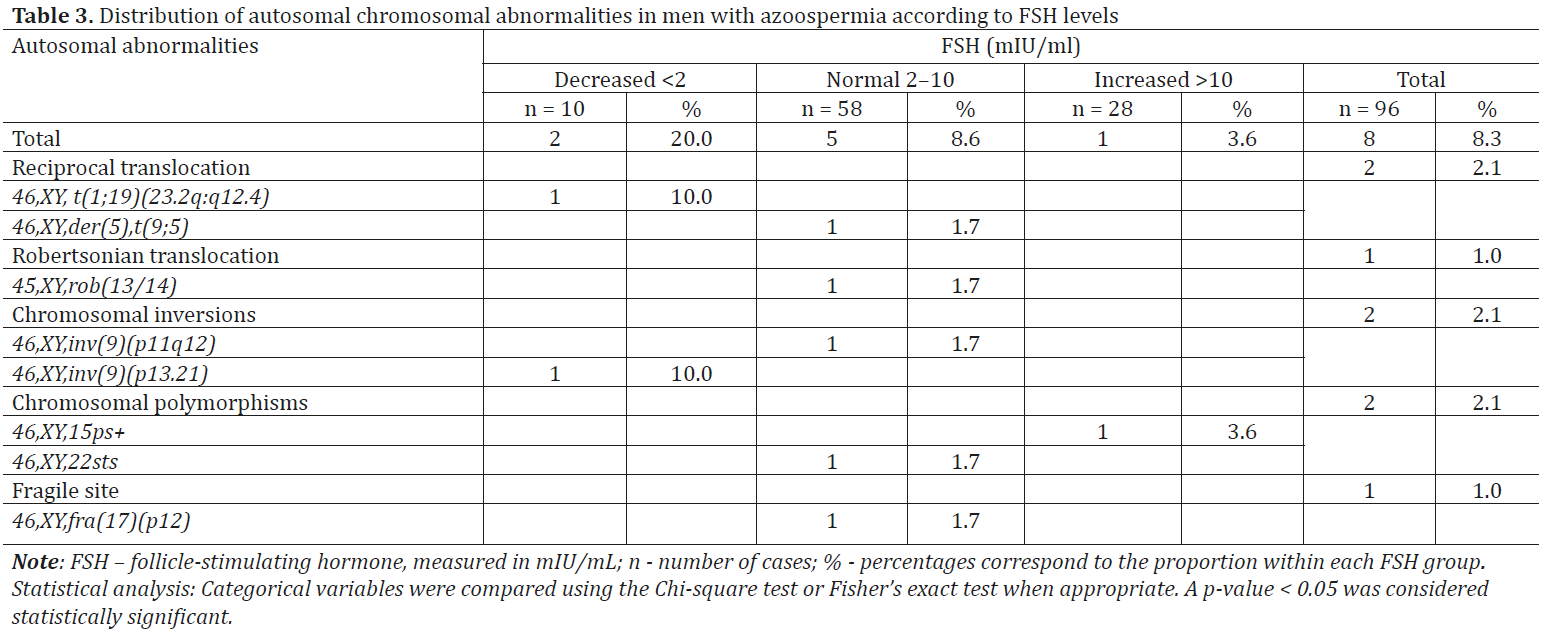
The most frequent autosomal chromosomal abnormalities detected were balanced chromosomal rearrangements, identified in 5 cases. Translocations were the most common balanced chromosomal abnormalities, found in 3.1% (n = 3) of the total cohort (n = 96). In the present study, among 96 azoospermic men, simple reciprocal translocations were detected in 2.1% (n = 2) of cases – specifically t(1;19) and t(9;5). In one case, a Robertsonian translocation involving the long arms of chromosomes 13 and 14 was identified, resulting in a karyotype with 45 chromosomes, present in all analyzed cells: rob(13;14) (Table 3, Fig. 8 and 9).
Chromosomal inversions were the most frequent balanced chromosomal rearrangements after translocations, identified in 2.1% (n = 2) of the total azoospermic men. Both patients exhibited pericentric inversions involving both arms of a chromosome from pair 9, detected in all analyzed cells: 46,XY,inv(9)(p11q12) and 46,XY,inv(9)(p13q21) (Table 3, Fig. 10, 11).
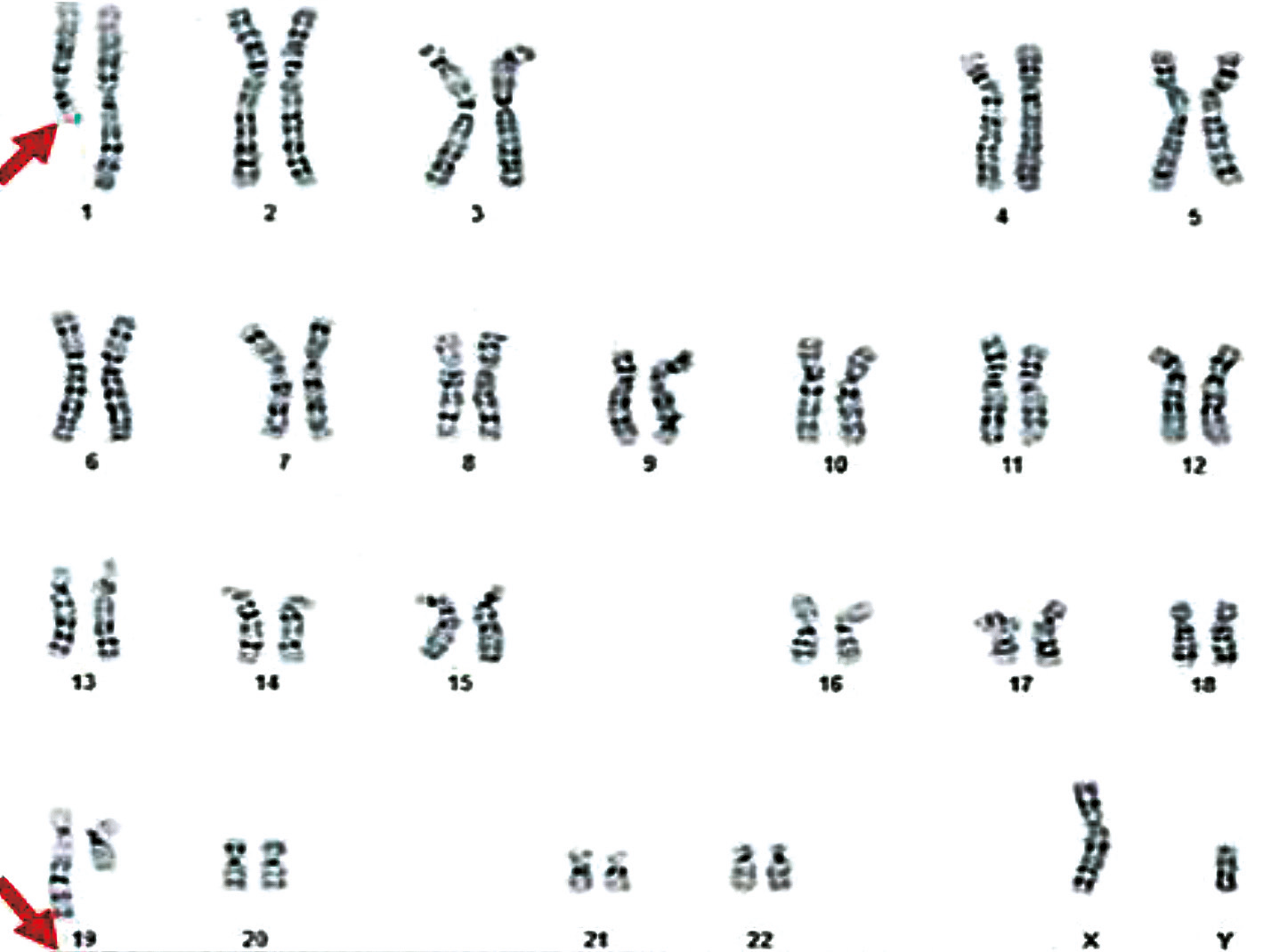 |
Fig. 8 Karyotype 46,XY,t(1;19)(q23.2;q13.4) of a 35-year-old patient Note: The karyotype 46,XY,t(1;19)(q23.2;q13.4) indicates the presence of a balanced translocation between the long arm (q) of chromosome 1 at region 23.2 and the long arm (q) of chromosome 19 at region 13.4, in a male with a normal chromosome number (46,XY). The chromosomes involved in the translocation are indicated by arrows. Metaphases counted: 33; Metaphases karyotyped: 10; Band resolution level: 650 |
|
Fig. 9 Karyotype 45,XY,rob(13;14)(q10;q10) of a 31-year-old patient Note: The karyotype 45,XY,rob(13;14)(q10;q10) indicates a Robertsonian translocation between chromosomes 13 and 14 at the centromeric regions (q10;q10) in a male with a total of 45 chromosomes instead of the normal 46. Chromosome banding technique: GTG; Metaphases counted: 33; Metaphases karyotyped: 10; Band resolution level: 600 |
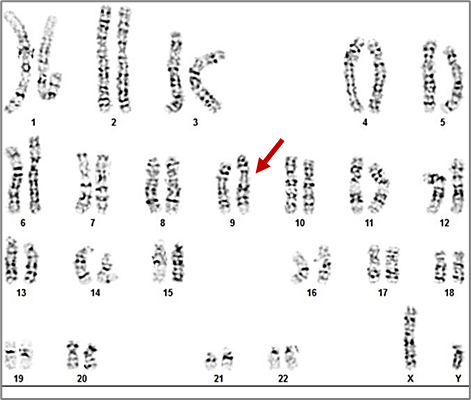
 |
Fig. 10 Karyotype 46,XY,inv(9)(p11q12) of a 35-year-old patient Note: The karyotype 46,XY,inv(9)(p11q12) represents a male with a normal chromosome number (46 chromosomes) who carries a pericentric inversion on chromosome 9. This inversion involves a segment between the short arm region p11 and the long arm region q12. Metaphases counted: 15; Metaphases karyotyped: 10; Band resolution level: 550-570 |
|
Fig. 11 Karyotype 46,XY,inv(9)(p13q21) of a 31-year-old patient Note: The karyotype 46,XY,inv(9)(p13q21) indicates a male with a normal chromosome number (46 chromosomes) who carries a pericentric inversion on chromosome 9. This inversion involves a segment between the short arm region p13 and the long arm region q21. |
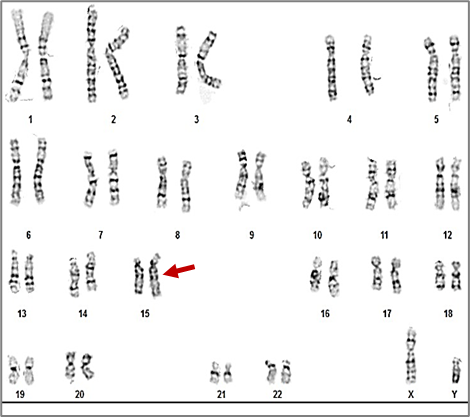
Chromosomal polymorphisms in the present study were identified in 2.1% (n = 2) of the 96 azoospermic men. One case exhibited an enlarged satellite region on the short arm of a chromosome from pair 15 (15ps+), and another on a chromosome from pair 21 (21ps+), observed in all analyzed cells (Table 3, Fig. 12, 13).
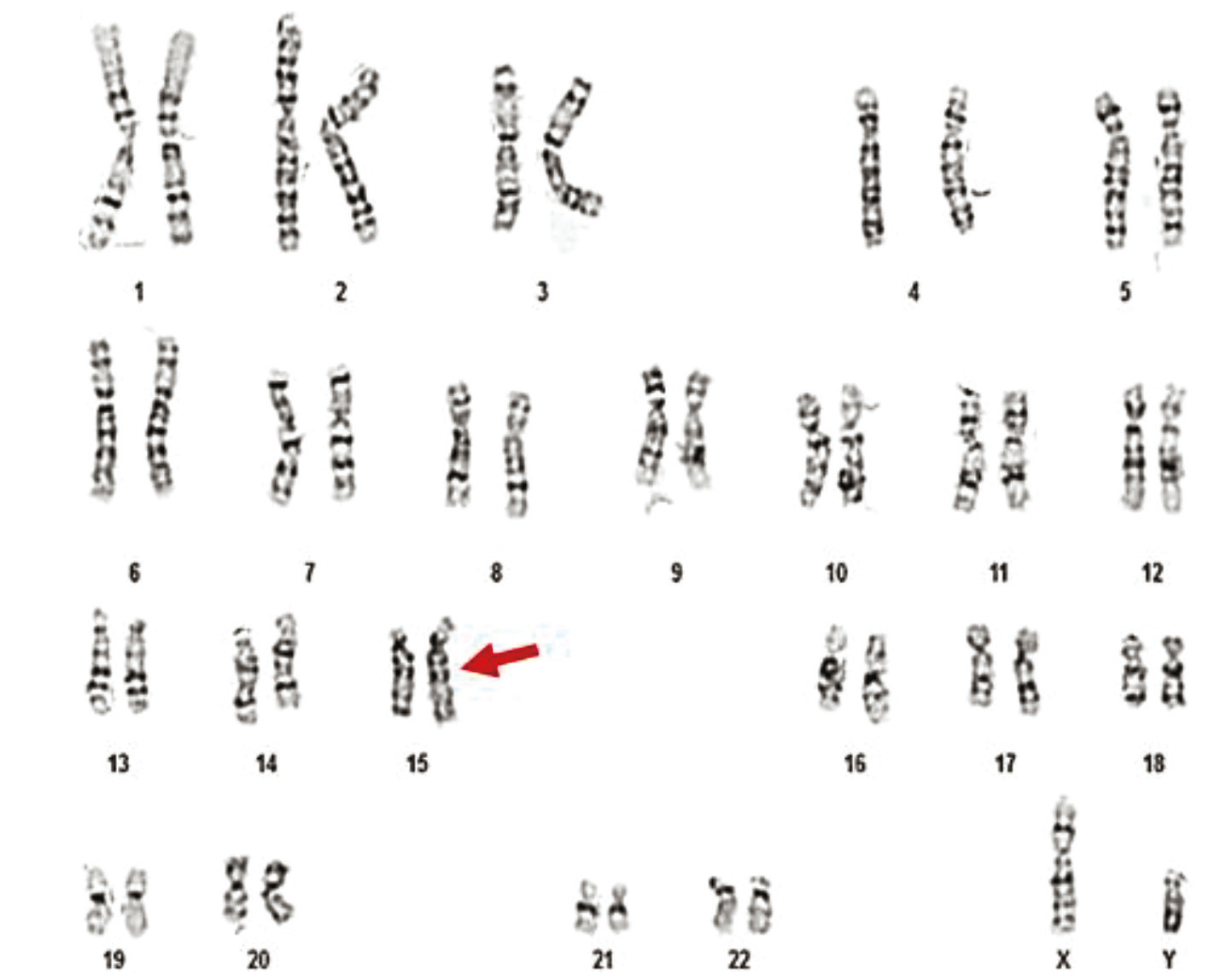 |
Fig. 12 Karyotype 46,XY,15ps+ of a 34-year-old patient Note: The image shows a standard human male karyotype with all 46 chromosomes. A chromosomal polymorphism is observed on chromosome 15, specifically on the short arm (p arm), and is noted as 15ps+. This indicates an enlargement of the satellite region of chromosome 15. The variation is considered benign and classified as a normal chromosomal polymorphism. |
|
Fig. 13 Karyotype 46,XY,22ps+ of a 32-year-old patient Note: The karyotype shows a standard male chromosomal complement with a total of 46 chromosomes, including one X and one Y chromosome (46,XY). A chromosomal polymorphism is observed on chromosome 22, specifically on the short arm (p arm), and is designated as 22ps+. This indicates an enlargement of the satellite region on the short arm of chromosome 22. The polymorphism is indicated in the image with a red arrow. |

Discussion
Men with azoospermia have the highest risk of being carriers of genetic abnormalities. The prevalence of chromosomal variations in men with azoospermia has been reported in the literature to range between 15% and 25% [11]. According to the cytogenetic analysis results from the present study, out of a total of 96 infertile men with azoospermia, 75% (n = 72) exhibited a normal 46,XY karyotype, while 25% (95% CI: 24.1 – 25.9; n = 24) showed numerical or structural chromosomal variations (Table 1, Fig. 1). It is worth noting that the prevalence of chromosomal abnormalities identified in this study (25.0%, 95% CI: 24.1 – 25.9) among azoospermic men is consistent with findings reported in other countries from the same geographical region. For example, similar or slightly higher rates have been observed in Ukraine (35.0%, 95% CI: 32.7 – 37.3), Romania (30.0%, 95% CI: 29.1 – 30.9), and Turkey (19.2%, range 18.2 – 20.2), while slightly lower frequencies were reported in Bulgaria (20.7%, range 19.3 – 22.1), Morocco (13.1%, 95% CI: 12.9 – 13.2), and China (14.7%, 95% CI: 14.6 – 14.8). Other studies from Western countries reported comparable or lower prevalence, such as the Netherlands (14.5%, 95% CI: 14.4 – 14.5), the USA (13.3%, 95% CI: 13.0 – 13.5), Italy (10.5%, 95% CI: 9.8 – 12.0), and India (11.0%, 95% CI: 10.8 – 11.3) [11-16].
It is well known that sex chromosome abnormalities are the most common chromosomal causes of infertility [17]. In our study, among the total number (n = 96) of patients with azoospermia, the prevalence of sex chromosome abnormalities was identified in 16.7% of cases (n = 16), while autosomal abnormalities were observed in 8.3% of cases (n = 8). Among the total cohort (n = 96) of azoospermic men investigated cytogenetically, 11 presented X disomy, representing 11.5% (Fig. 1, Tables 1, 2). The results of our research are consistent with data from the literature, which report a similarly high frequency of KS among azoospermic men, ranging from 10% to 15%.
The phenotype of patients with KS is highly heterogeneous, being influenced by both the karyotype type and the age of the patient. During childhood, KS often remains undiagnosed due to its non-specific clinical presentation. Clinical manifestations typically become apparent in adulthood, with the onset of gynecomastia, hypogonadism, and infertility [18]. All patients in the present study were diagnosed in adulthood due to infertility. The cytogenetic variants identified in our study support the data in the literature, which report a high incidence (80-90%) of the classical 47,XXY form of KS, while mosaic forms are described in approximately 20% of cases. The 47,XXY aneuploidy is the most common sex chromosomal abnormality, with an incidence ranging from 1 in 500 to 1 in 1,000 live male births. Other cytogenetic variants involving X polysomies are rare: 48,XXXY (1 in 50,000) and 49,XXXXY (1 in 85,000) [19-20].
The identification of the cytogenetic variant in KS has significant clinical importance, as the severity of the clinical phenotype is directly proportional to the number of supernumerary X chromosomes. The phenotype depends on the extent of genetic imbalance caused by genes that escape X inactivation, androgen deficiency, and the sensitivity of androgen receptors. A greater degree of genetic imbalance, androgen deficiency, and impaired androgen receptor sensitivity correlates with a more severe phenotype. The clinical presentation of KS progressively worsens with each additional X chromosome [19, 21-23].
Among the total cohort (n = 96) of azoospermic men who underwent cytogenetic evaluation, one case was diagnosed with Y chromosome disomy, with a frequency of 1.04% (Table 2). Cytogenetic analysis revealed the presence of an extra Y chromosome in all analyzed cells, with a karyotype of 47,XYY (Fig. 4).
The 47,XYY syndrome is relatively common, occurring in approximately 1 in 1,000 live male births, making it the second most frequent sex chromosome abnormality after Klinefelter syndrome. However, up to 85% of XYY males remain undiagnosed. This was consistent with our case, as the 35-year-old male showed no clinical signs suggestive of a chromosomal abnormality, and the cytogenetic investigation was prompted solely by infertility due to azoospermia [24, 25].
According to the literature, most individuals with a 47,XYY karyotype exhibit normal spermatogenesis, while a minority may present varying degrees of spermatogenic impairment, ranging from normal to azoospermia [8, 25]. In the presented case, the patient had azoospermia, which may be explained by abnormal chromosomal pairing during spermatogenesis due to the presence of an additional Y chromosome.
FSH levels are typically elevated in such patients as a response to inadequate spermatogenesis, while testosterone levels are usually within normal limits or slightly increased. In this patient, both FSH and testosterone levels were within normal ranges.
Studies comparing the semen parameters of fertile and infertile men with XYY syndrome have shown that the majority of spermatozoa in these individuals have a normal karyotype.
In one azoospermic male from the total cohort (n = 96), a 46,XX karyotype was identified (1.04%) (Table 3, Fig. 5). The occurrence of 46,XX males in the general population is extremely rare (1 in 20,000), with a reported prevalence of approximately 0.9% among azoospermic men and 1–3% among normozoospermic men [9].
Following cytogenetic evaluation (46,XX karyotype), FISH analysis revealed two signals for the X chromosome and one signal for the Y chromosome (SRY, Yp11.32), and molecular genetic testing confirmed the presence of the SRY gene. Based on these findings, the diagnosis of testicular disorder of sex development (DSD), sex reversal, 46,XX male, also known as de la Chapelle syndrome, was established.
De la Chapelle syndrome is a form of sex reversal characterized by a female karyotype discordant with a male phenotype. Two categories have been described in the literature: SRY-positive cases, accounting for approximately 80% of patients, and SRY-negative cases, representing the remaining 20%. The present case falls into the SRY-positive category, as confirmed by multiplex PCR molecular testing and FISH analysis, which demonstrated the presence of the SRY gene translocated onto one of the X chromosomes [26, 27].
In one azoospermic patient, a 45,X/46,XY mosaic karyotype was identified (Table 3), a rare condition with a reported frequency of approximately 1 in 15,000 live births [10]. The clinical significance of 45,X/46,XY mosaicism is controversial in the literature and presents a major clinical challenge, as it is associated with a broad spectrum of manifestations ranging from infertility and ambiguous genitalia to phenotypically normal males [28-31].
According to the cytogenetic analysis, the patient with 45,X/46,XY mosaicism exhibited two distinct cell lines: 45,X (X monosomy) in 20% of the cells and a normal male 46,XY karyotype in 80% of the cells. This distribution explains the phenotypically male appearance and the absence of any suggestive clinical signs. The indication for karyotyping was a severely impaired semen analysis and infertility, which were diagnosed at a late age (46 years).
Y chromosome microdeletions represent the second most common genetic cause of impaired spermatogenesis in infertile men, following Klinefelter syndrome. In this study, two cases with structural variations of the Y chromosome were identified: 46,X,del(Y)(q11.21) (Y ≤ 21) and 46,XYqh+ (Y ≥ 18).
The prevalence of Y chromosome microdeletions is estimated to be approximately 1 in 2,000 to 1 in 3,000 men [32]. In this study, such a deletion was detected in 1 out of 96 azoospermic men, with a prevalence of 1.04% (Table 2). In a 36-year-old patient, an abnormal male karyotype was identified: 46,X,del(Y)(q11.21), indicating an unbalanced structural alteration (deletion) involving a portion of the long arm of the Y chromosome (region Yq11.21–qter) in all analyzed cells.
Polymorphic variants of the Y chromosome (Yqh+) have been reported in several studies concerning male infertility, particularly among men with azoospermia and severe oligozoospermia. In the present study, a chromosomal Y polymorphism was detected in one case with 46,XYqh+(Y ≥ 18), corresponding to a prevalence of 1.04% in the total sample (n = 96) (Table 2). Yqh+ represents a variation in the constitutive heterochromatin region of the Y chromosome, which cannot directly explain spermatogenic disorders. However, this topic remains controversial due to the potential role of heterochromatin, whose clinical significance has not yet been fully elucidated. The Y(≥18) variant results from excessive duplication of the DY21 sequence typical of the heterochromatic region of the Y chromosome. This may lead to mitotic errors, gene expression dysregulation, and impaired cellular differentiation, potentially resulting in gestational issues [27].
Autosomal chromosome variations have also been described in the literature among infertile patients, often without phenotypic manifestations. In the current study, among 96 azoospermic men, such variations were found in 8.3% (n = 8) of cases (Table 3).
The most frequent autosomal findings were balanced chromosomal rearrangements, found in 5 cases. Among these, translocations were the most common type of balanced rearrangement. In the current study, they were detected in 3.1% (n = 3) of the total cohort (n = 96). Balanced chromosomal translocations involve breaks in two chromosomes and the abnormal rearrangement of chromosomal fragments, resulting in the exchange of genetic material without any loss. This typically explains why most translocation carriers have a normal phenotype. Azoospermia is largely attributed to the possibility that one of the breakpoints disrupts a gene directly involved in spermatogenesis, potentially leading to its arrest or incomplete development.
In this study, among 96 azoospermic men, simple balanced translocations were detected in 2.1% (n = 2) of cases – t(1;19) and t(9;5) (Fig. 8). In one case, a Robertsonian translocation involving 45 chromosomes was identified, affecting the long arms of chromosomes 13 and 14 in all analyzed cells: rob(13;14) (Fig. 9). According to the literature, this translocation between chromosomes 13 and 14 is the most common, followed by translocation between chromosomes 14 and 21 [33].
Chromosomal inversions are the second most common type of balanced chromosomal rearrangement after translocations. This finding is also supported by the current study, in which inversions were identified in 2.1% (n = 2) of cases among azoospermic men (Table 3, Fig. 10, 11). The probability that inversion carriers produce abnormal gametes due to meiotic crossover events ranges from 1% to 10%. As a result of homologous chromosome recombination within the inversion loop, four types of gametes can be produced: one normal, one with the inversion, and two partially duplicated or deleted [34].
Conclusions
The detection of an abnormal karyotype is highly important for comprehensive and effective genetic counseling. This process should include detailed information about the specific type of chromosomal abnormality or polymorphism, its clinical relevance, potential inheritance patterns, genetic risk to offspring, and available options for prenatal diagnosis.
Genetic counseling guides infertile couples in making informed decisions regarding medically assisted reproduction. Therefore, cytogenetic and molecular genetic screening remains a valuable practice for accurate diagnosis, assessment, prognosis, and successful treatment.
Competing interests
None declared.
Authors’ contribution
SR conceived conceptualization, methodology, data collection, analysis and interpretation, writing – original draft preparation. SR, EC and VM analyzed the result. MS, VR and AM – supervision on differential diagnosis data. MS and SC – research coordinator, conceived writing review and editing, validation. The authors read and approved the final version of the manuscript.
Patient consent
Obtained
Ethics approval
This study was approved by the Research Ethics Committee of Nicolae Testemițanu State University of Medicine and Pharmacy, Protocol No 48 of April 12, 2018.
Acknowledgments and funding
No external funding.
Provenance and peer review
Not commissioned, externally peer review.
Authors’ ORCID IDs
Stela Racoviță – https://orcid.org/0000-0002-0900-0096
Veaceslav Moşin– https://orcid.org/0000-0002-1209-525X
Svetlana Capcelea – https://orcid.org/0000-0003-2656-8254
Ana Mișina – https://orcid.org/0000-0001-6248-0319
Vasile Racoviță – https://orcid.org/0009-0001-9898-4831
Elena Chesov – https://orcid.org/0000-0001-8942-2282
Mariana Sprincean – https://orcid.org/0000-0002-3619-1924
References
American Urological Association, American Society for Reproductive Medicine. Report on evaluation of the azoospermic male. Fertil Steril. 2006;86(5 Suppl 1):S210-5. doi: 10.1016/j.fertnstert.2006.08.030.
Wosnitzer M, Goldstein M, Hardy MP. Review of azoospermia. Spermatogenesis. 2014;4(1):e28218. doi: 10.4161/spmg.28218.
Samli H, Samli MM, Solak M, Imirzalioglu N. Genetic anomalies detected in patients with non-obstructive azoospermia and oligozoospermia. Arch Androl. 2006;52(4):263-7. doi: 10.1080/01485010600664032.
Lee JY, Dada R, Sabanegh E, Carpi A, Agarwal A. Role of genetics in azoospermia. Urology. 2011;77(3):598-601. doi: 10.1016/j.urology.2010.10.001.
Esteves SC, Hamada A, Kondray V, Pitchika A, Agarwal A. What every gynecologist should know about male infertility: an update. Arch Gynecol Obstet. 2012;286(1):217-29. doi: 10.1007/s00404-012-2274-x.
Pylyp LY, Spinenko LO, Verhoglyad NV, Zukin VD. Chromosomal abnormalities in patients with oligozoospermia and non-obstructive azoospermia. J Assist Reprod Genet. 2013;30(5):729-32. doi: 10.1007/s10815-013-9990-4.
Donker RB, Vloeberghs V, Groen H, Tournaye H, van Ravenswaaij-Arts CMA, Land JA. Chromosomal abnormalities in 1663 infertile men with azoospermia: the clinical consequences. Hum Reprod. 2017;32(12):2574-80. doi: 10.1093/humrep/dex307.
Racoviță S, Moșin V, Hadjiu S, Poneatenco D, Revenco N, Sprincean M, et al. Sindromul 47,XYY asociat cu infertilitatea masculină: raport de caz clinic [47,XYY syndrome associated with male infertility: clinical case report]. Bull Perinatol (Chisinau). 2020;86(1):112-5. Romanian.
Racoviță S, Moşin V, Capcelea S, Poneatenco D, Boiciuc K, Sprincean M. Bărbat 46,XX, raport de caz clinic [46,XX male: clinical case report]. Bull Perinatol (Chisinau). 2019;83(2):104-7. Romanian.
Racoviță S, Sprincean M, Moşin V, Capcelea S, Hadjiu S, Revenco N. 45,X/46,XY la bărbat cu infertilitate: raport de caz clinic [45,X/46,XY in men with infertility: clinical case report]. In: [Proceedings of the 5th scientific national conference „Interdisciplinary management of children”]. Chisinau; 2022. p. 145-149. Romanian.
Cozaru GC, Butnariu LI, Gorduză EV. Genetic counselling in reproductive disorders. Procedia Soc Behav Sci. 2012;33:213-7. doi: 10.1016/j.sbspro.2012.01.114.
Pylyp LY, Spinenko LO, Verhoglyad NV, Zukin VD. Chromosomal abnormalities in patients with oligozoospermia and non-obstructive azoospermia. J Assist Reprod Genet. 2013;30(5):729-32. doi: 10.1007/s10815-013-9990-4.
Kovacheva K, Kotsev R, Konova E, Rilcheva V, Kamburova Z, Simeonova M. Chromosomal abnormalities and Y chromosome microdeletions in Bulgarian male with azoospermia or severe oligospermia. J Int Med Assoc Bulgaria. 2018;24(4):2217-22. doi: 10.5272/jimab.2018244.2217.
Bellovits O, Rusz A , Romics I, Csonka E, Hadlaczky G, Sótonyi P et al. Chromosomal aneuploidy in azoospermic men. Int J Hum Genet. 2006;6(2):171-6. doi: 10.1080/09723757.2006.11885959.
Naasse Y, Charoute H, El Houate B, Elbekkay C, Razoki L, Malki A, et al. Chromosomal abnormalities and Y chromosome microdeletions in infertile men from Morocco. BMC Urol. 2015;15(1):6. doi: 10.1186/s12894-015-0089-3.
Christofolini DM, Mafra FA, Neto RP, Saab De Almeida Barros RA, Amaro Dos Santos A, Peluso C, et al. Correlation between chromosomal variants and male infertility in a population of Brazilian infertile men. Reprod Syst Sex Disord. 2012;1(1):1-6. doi: 10.4172/2161-038X.1000105.
Racovița S, Mosin V, Capcelea S, Misina A, Sprincean M. Chromosomal abnormalities in men with azoospermia. Mold Med J. 2021;64(1):50-5. doi: 10.5281/zenodo.4527139.
Sprincean M, Barbova N, Halabudenco E, Mişina A, Samoilenco T. Aspecte ale diagnosticului timpuriu şi particularităţile polimorfismului clinic şi citogenetic în sindromul Klinefelter [Early diagnosis and clinical/cytogenetic polymorphism in Klinefelter syndrome]. In: Scientific annals of Nicolae Testemitanu SUMPh. Chisinau: Medicina; 2013. Vol. 5. p. 426-433. Romanian.
Tüttelmann F, Gromoll J. Novel genetic aspects of Klinefelter’s syndrome. Mol Hum Reprod. 2010;16(6):386-95. doi: 10.1093/molehr/gaq019.
Racoviță S, Capcelea S, Moşin V, Halabudenco E, Mişina A, Samoilenco T, et al. Variații cromozomiale la bărbații infertili [Chromosomal variations in infertile men]. Bull Acad Sci Mold. Med Sci. 2020;(3):78-82. Romanian.
Racoviță S, Moșin V, Hadjiu S, Revenco N, Călcîi C, Cuznetz L, et al. Neurogenetic aspects in men with Klinefelter syndrome. Mold Med J. 2021;64(Suppl Neuro Congress):55.
Racoviță S, Sprincean M, Moşin V, Hadjiu S, Barbova N, Halabudenco E, et al. Polimorfism clinic și variații citogenetice în infertilitatea masculină cauzată de Sindromul Klinefelter [Clinical polymorphism and cytogenetic variations in male infertility caused by Klinefelter syndrome]. Bull Acad Sci Mold. Med Sci. 2018;(1):44-8. Romanian.
Racoviță S, Sprincean M, Hadjiu S. Neurological impairment and cytogenetic variations in Klinefelter syndrome. Eur J Neurol. 2022;29(Suppl 1):906. doi: 10.1111/ene.15467.
Flannigan R, Schlegel PN. Genetic diagnostics of male infertility in clinical practice. Best Pract Res Clin Obstet Gynaecol. 2017;44:26-37. doi: 10.1016/j.bpobgyn.2017.05.002.
Kim IW, Khadilkar AC, Ko EY, Sabanegh ES. 47,XYY syndrome and male infertility. Rev Urol. 2013;15(4):188-96.
Akinsal EC, Baydilli N, Demirtas A, Saatci C, Ekmekcioglu O. Ten cases with 46,XX testicular disorder of sex development: single center experience. Int Braz J Urol. 2017;43(4):770. doi: 10.1590/S1677-5538.IBJU.2016.0505.
Dominguez-Lopez M, Gonzalez-Molero I, Esteva I, Gonzalo-Marin M, MSoledad Ruiz de A, Federico S. Clinical case report: male patient with SRY-positive 46,XX testicular disorder of sex development. Endocr Abstr. 2013;32:P288. doi: 10.1530/endoabs.32.P288.
Yahaya TO, Oladele EO. Anyebe D, Obi C, Bunza MDA, Sulaiman R, Liman UU. Chromosomal abnormalities predisposing to infertility, testing, and management: a narrative review. Bull Natl Res Cent. 2021;45:65. doi: 10.1186/s42269-021-00523-z.
Racoviță S, Patrașcu A, Capcelea S, Mișina A, Samoilenco T, Sprincean M. Cytogenetic study in male infertility associated with azoospermia and severe oligozoospermia. In: 11th International Congress of Geneticists and Breeders from the Republic of Moldova, 2021 Jun 15-16; Chisinau: Abstract book. Chișinău: USM; 2021. p. 32. doi: 10.53040/CGA11.2021.015.
Tosson H, Rose SR, Gartner LA. Description of children with 45,X/46,XY karyotype. Eur J Pediatr. 2012;171(3):521-9. doi: 10.1007/s00431-011-1600-9.
Flannigan RK, Chow V, Ma, S, Yuzpe A. 45,X/46,XY mixed gonadal dysgenesis: a case of successful sperm extraction. Can Urol Assoc J. 2014;8(1-2):E108-10. doi: 10.5489/cuaj.1574.
Özdemir TR, Özyılmaz B, Çakmak Ö, Kaya ÖÖ, Köse C, Kırbıyık Ö, et al. Evaluation of chromosomal abnormalities and Y-chromosome microdeletions in 1696 Turkish cases with primary male infertility: a single-center study. Turk J Urol. 2020;46(2):95-100. doi: 10.5152/tud.2019.19156.
Kate UV, Pokale YS, Jadhav AM. Gangane SD. Chromosomal aberrations and polymorphic evaluation in males with primary infertility from Indian population. J Clin Diagn Res. 2014;8(10):SC01-6. doi: 10.7860/JCDR/2014/8644.4933.
Xie X, Li F, Tan W, Tang J. Analysis of the clinical features of pericentric inversion of chromosome 9. J Int Med Res. 2020;48(9):1-9. doi: 10.1177/0300060520957820.