
Introduction
The DiGeorge syndrome (DGS) is a collection of signs and symptoms associated with the faulty development of the pharyngeal pouch system. Most cases are caused by a heterozygous chromosomal deletion at 22q11.2 discovered in the 1980s. The 22q11.2 chromosomal region contains approximately 30–40 genes that are critically involved in the embryologic formation of the third and fourth pharyngeal arches, thereby influencing the development of the cardiac outflow tract, great vessels, thymus, parathyroid glands, and craniofacial structures [1-4].
A comprehensive study conducted on a large population has shown that heterozygous chromosome 22q11.2 deletions are relatively widespread in the general population, establishing 22qDGS as the most common microdeletion syndrome. According to this study, approximately 1 in 5950 live births exhibited a deletion in this chromosomal region, with 83 percent of these infants also presenting a cardiac defect [5]. Another population-based study reported an incidence rate of 22qDGS as high as 1 in 4000 live births [6, 7]. Its frequency is 2.8-5% for congenital cardiac malformations. In conotruncal defects, this rate is reported as 10-19.4% in various studies [1,8]. The vast majority of DiGeorge syndrome cases (approximately 90%) occur spontaneously, although autosomal dominant inheritance has been reported in a minority of patients (8–28%) [7, 9].
The deletion of chromosome 22q11.2 presents a wide spectrum of clinical features, including heart and major blood vessel defects, underdevelopment of the parathyroid glands, deficiency in parathyroid hormone, hypocalcemia, seizures, aplasia and hypoplasia of the thymus, immunological disorders, growth hormone deficiency, anomalies of the facial skeleton and structure of the larynx, pharynx, trachea, inner ear, esophagus, teeth, kidney malformations, gastrointestinal and central nervous system anomalies, delayed speech and motor development, hyperactivity syndrome, and schizophrenia [10, 11].
In a study conducted on a group of postmortem-diagnosed DGS patients, alongside severe immune system alterations, these children exhibited various stigmata of dysmorphogenesis, such as low-set auricles, hypotelorism or hypertelorism, micrognathia, high palatal vault, cleft palate, saddle nose, etc., as well as various congenital malformations [12, 13].
In DGS, immunological defects vary from extended and recurring sinopulmonary infections (often referred to as partial DGS) to congenital athymia (a phenotype resembling severe combined immunodeficiency [SCID], termed complete DGS). The level of immunodeficiency severity correlates with the extent of thymic functionality [14].
Individuals with DGS exhibit increased susceptibility to autoimmune conditions, such as autoimmune cytopenia and juvenile idiopathic arthritis [15].
The objective of this study was to outline the clinical characteristics observed in a cohort of genetically confirmed DGS patients, with the aim of enhancing awareness and comprehension of DGS.
Material and methods
This descriptive observational study was conducted in the Human Molecular Genetics Laboratory and the Department of Pediatrics of the Institute of Mother and Child and included 10 consecutive patients investigated for suspected 22q11.2 syndrome. Participants were recruited based on clinical criteria formulated by Tobias et al. [16, 17], according to which inclusion was possible in the presence of a major conotruncal heart defect (such as tetralogy of Fallot, interrupted aortic arch, truncus arteriosus, or major aortopulmonary collaterals), two or more core features – characteristic facial anomalies, non-conotruncal heart defects, developmental delay or learning disabilities, hypocalcemia, immunodeficiency, or thymic hypoplasia – or a core criterion associated with complementary manifestations, such as long and thin fingers, short stature, hypotonia, palatal malformations (cleft palate, velopharyngeal insufficiency, difficulty swallowing), renal abnormalities, family history of congenital heart disease, or psychiatric disorders, especially bipolar disorder. For each patient, clinical data were collected in a standardized manner. The assessment of height and weight development was performed using SD scores and age-appropriate growth charts, and recurrent respiratory morbidity was assessed according to criteria described in the specialized literature [18].
All patients underwent a complete interdisciplinary evaluation, which included cardiological examination with Doppler echocardiography to identify conotruncal or non-conotruncal malformations, endocrinological evaluation consisting of the determination of total and ionized serum calcium and thyroid hormone levels (ELISA method), as well as ENT examination (including audiogram). The neurological examination was completed by neuropsychological evaluation, and the immunological profile included complete blood count, absolute lymphocyte count, platelet count, serum immunoglobulin levels (ELISA method), and lymphocyte subset analysis (performed by flow cytometry).
The genetic diagnosis of the 22q11.2 deletion was established using high-accuracy molecular methods, according to internally validated protocols, including multiplexed QF-PCR using FAM channels for internal controls, VIC/HEX for target regions, and LIZ for the size marker [13, 19, 20], as well as fluorescence in situ hybridization (FISH), chromosomal microarray analysis (CMA), or droplet digital PCR (ddPCR) in cases that required additional confirmation. All analyses were performed under standardized conditions to ensure reproducibility of the results.
Data processing and descriptive statistical analysis were performed using Microsoft Excel (Microsoft Corporation, Excel, version 2010, WA, USA) for data organization, and IBM SPSS Statistics version 26.0 (IBM Corp., Armonk, NY, USA), which was used to investigate the descriptive characteristics of the entire cohort.
The study was approved by the Research Ethics Committee of the Nicolae Testemițanu State University of Medicine and Pharmacy (No. 13/15.03.2019), and informed consent was obtained from all legal guardians of the participating minors.
Results
The mean age of patients was 9.3 years (2–30 years) at the time of the study and 74.6 months (3 months – 28 years) at diagnosis. In the study group, 30% of the patients were male, while 70% were female. Demographic and clinical features of the cohort are described in Table 1.
Table 1. Demographic and clinical features of patients with 22q11.2 deletion syndrome | |||||
Patient | Current age (years) / gender | Age of onset (months) | Cardiac defect | Other abnormalities | Family history |
1 | 5/female | 3 | Common truncus | Facial dysmorphism Growth retardation | None |
2 | 4/male | 48 | Tetralogy of Fallot | Facial dysmorphism | Great-grandmother and uncle with cardiac malformation |
3 | 8/female | 24 | Tetralogy of Fallot | Facial dysmorphism Growth retardation Psychomotor, cognitive and language delay, attention-deficit hyperactivity Upper and lower respiratory tract infections, otitis Renal agenesis Swallowing difficulty | Father with DGS |
4 | 30/male | 28 years | Unspecified | Facial dysmorphism | Father of P3 |
5 | 8/female | 72 | None | Upper and lower respiratory tract infections | None |
6 | 5/female | 10 | Tetralogy of Fallot | Facial dysmorphism Growth retardation Laryngomalacia | None |
7 | 9/female | 60 | Patent foramen ovale (PFO) | Facial dysmorphism Cognitive and language delay | None |
8 | 2/female | 6 | Tetralogy of Fallot | Facial dysmorphism Growth retardation | None |
9 | 2/female | 7 | Right aortic arch | Upper and lower respiratory tract infections Facial dysmorphism Growth retardation | None |
10 | 20/male | 15 years | Tetralogy of Fallot | Facial dysmorphism Upper and lower respiratory tract infections | None |
Note: DGS - DiGeorge syndrome; PFO - patent foramen ovale; P3- patient number 3 | |||||
Most cases were sporadic, with only 2 patients having a history of DGS (n=1) or close relatives with cardiac malformations (n=1). During the neonatal period, subjects P3 and P9 experienced complications such as seizures, hypotonia, and sucking difficulties.
Cardiac malformations were the most common manifestation, accounting for 90% of cases (n=9). Among these, 77.7% exhibited conotruncal anomalies (5 had tetralogy of Fallot, 1 had right aortic arch, 1 – common truncus), while 33.3% had non-conotruncal anomalies. All patients with cardiac malformations required surgical correction, during which thymic hypoplasia was described in all cases.
Nine of ten patients (90%) exhibited facial dysmorphism, with microstomia being the most prevalent feature (9/10), followed by micrognathia (4/10), dental anomalies (1/10), hypertelorism (3/10), and low-set ears (3/10).
Forty percent of the patients presented recurrent infections, predominantly in the form of pneumonia. Following surgical correction, the number of infections decreased in most patients.
ENT anomalies were recorded in 20% of the patients, with one presenting laryngomalacia and one exhibiting swallowing difficulties and palatal anomalies. Other alterations observed in patients with DGS included speech delay in 2 patients and developmental delay in 2 patients. Regarding congenital renal conditions, renal agenesis was identified in P3.
Weight was within normal percentiles for the entire group, but a delay in height growth was noted. Most of the cohort experienced a decrease in height (between -1 and -2 SD), particularly during childhood. There were no instances of short stature (height < -2 SD) recorded. Subject P4 was excluded due to a lack of reported measurements. P3 presented with neonatal hypocalcemia.
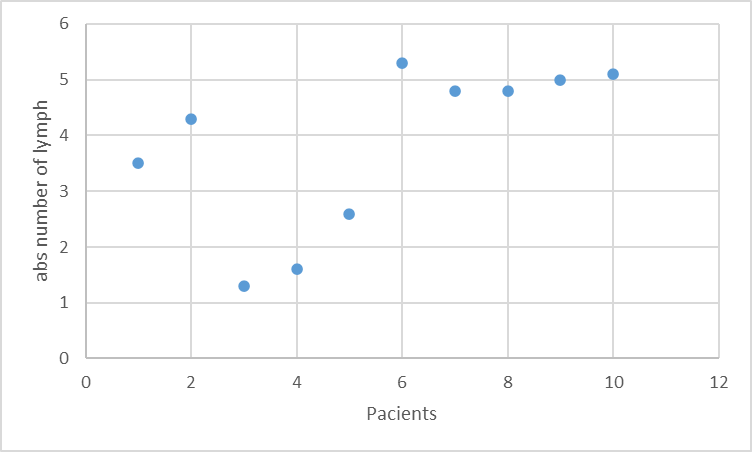
From the immunological characteristics, lymphopenia was recorded in 20% of patients (Fig.1), with a mean value of 3.83 ± 1,48; low platelet counts (100-150,000) were observed in 2 patients. IgA was below normal levels in 1 patient. Elevated levels of IgE were noted in 2 patients.
|
Fig.1 The lymphocyte count in the DGS group Note: Absolute lymphocyte count was calculated by multiplying the percentage of lymphocytes by the total leukocyte count (×10⁹/L). Lymphopenia was defined as less than 3 ×10⁹/L for children under 3 years of age and less than 1.5 ×10⁹/L for those over 3 years of age. |
Taking into account the presence of recurrent infections, predominantly pneumonia, patients underwent chest X-rays, which consistently showed the absence of a thymic shadow, a characteristic sign of DGS.
Discussion
The present study provided a clinical characterization of a group of patients with DGS. Considering the high clinical heterogeneity and the state of underdiagnosis in DGS, elaborating upon the clinical features of DGS may be helpful for improving timely diagnoses and diagnosis rates. In this group of patients, typical characteristics associated with 22q11.2DS were identified. These included congenital heart defects, distinct facial features, recurrent infections, and abnormalities in the immune system.
All patients included in the study were diagnosed with 22q11.2 deletion. In approximately 90% of cases, the 22q11.2 deletion arises de novo, while in 6–25% of patients it is inherited in an autosomal dominant manner [1, 21]. In line with previously published data, our cohort included one patient (10%) with a paternally inherited deletion, as well as one patient with a family history of congenital cardiac anomalies. These findings underscore the importance of comprehensive genetic counseling for affected families, including a clear discussion of recurrence risk and available prenatal diagnostic options [1].
As anticipated, congenital heart defects were a prominent feature, presenting in 90% of our patients. This prevalence aligns with prior findings, where 75% of individuals with the 22q11.2 deletion exhibit cardiac anomalies, predominantly conotruncal defects or aortic arch anomalies [1, 22], or 49–83%, according to other sources [23]. Among these, tetralogy of Fallot and interrupted aortic arch are the most common (27-62.3%; 14-53%, respectively). Transposition of the great arteries is infrequent. Non-conotruncal structural cardiac anomalies are also documented, with deletions potentially implicated in 5% of all newborns with cardiac defects [1, 24].
Another prominent characteristic of DiGeorge syndrome observed in our group was recurrent infections, which occurred in 40% of the patients. Jawad et al. and Oskarsdottir et al. reported recurrent infections in 60–65% of their study populations [25, 26]. Subsequent studies by Giardino et al., Cancrini et al., and Nissan et al. revealed even higher rates of recurrent infections in individuals with DiGeorge syndrome (64%, 54%, and 78%, respectively) [27-29]. These disparities between our cohort and previous studies may be attributed to the focus on other criteria, such as the documentation of recurrent infections.
Distinctive facial dysmorphic characteristics, including microstomia, narrow palpebral fissures, and micrognathia, were noted in all study patients. Additionally, less common traits, such as low-set ears, dental anomalies, and hypertelorism were also observed. The findings may differ based on race and ethnicity [30, 31]. In this study, the majority of patients with the 22q11.2 deletion exhibited the characteristic facial features of the syndrome. This underscores the significance of visual inspection in clinical assessment.
In our cohort, 20% of patients exhibited speech and developmental delays, which is lower than the prevalence reported in earlier studies [32].
Hematologic abnormalities, particularly thrombocytopenia and lymphopenia, have been extensively documented in individuals with DiGeorge syndrome in the literature. Thrombocytopenia was described in some cases as having an autoimmune etiology, but in most cases, its etiology is unknown [32, 33].
No severe immunodeficiency resembling leaky SCID was observed in our cohort. The immunological abnormalities detected may reflect thymic hypoplasia, commonly reported in 22q11.2DS [28, 34].
Conclusions
In conclusion, DiGeorge syndrome is a condition that impacts various organ systems. Prompt detection of this syndrome is key to proper care. Given the diverse array of symptoms associated with DiGeorge syndrome, physicians should be knowledgeable about both typical and less common characteristics of the syndrome to facilitate optimal treatment and potentially enable early diagnosis. This marks the initial report of DGS patients in our country. Despite the relatively high frequency of DGS, only a small number of patients with a confirmed diagnosis are currently under follow-up.
Competing interests
None declared.
Authors’ contributions
TC conceived the conceptualization, methodology, data collection, analysis, and interpretation, and was responsible for writing – original draft preparation; VS, DA, and LM – coordination of genetic analysis; SS – research coordination, conception, review writing, editing, and validation. All authors read and approved the final manuscript.
Acknowledgements
No external funding.
Ethics approval
The study protocol was approved by the Research Ethics Committee of the Nicolae Testemițanu State University of Medicine and Pharmacy (No.13 from 15.03.2019).
Provenance and peer review
Not commissioned, externally peer-reviewed.
Authors’ ORCID IDs
Cristina Tomacinschii – https://orcid.org/0000-0001-6351-5659
Victoria Sacara –https://orcid.org/0000-0001-9200-0494
Alexandr Dorif – https://orcid.org/0000-0003-4269-4066
Marodi Laszlo – https://orcid.org/0000-0002-9692-3402
Svetlana Sciuca – https://orcid.org/0000-0003-1091-9419
References
Celep G, Oğur G, Günal N, Baysal K. DiGeorge syndrome (Chromosome 22q11. 2 deletion syndrome): A historical perspective with review of 66 patients. Journal of Surgery and Medicine. 2019 Jan 27;3(1):58-63.
Grati FR, Molina Gomes D, Ferreira JC, Dupont C, Alesi V, Gouas L, et al. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenat Diagn. 2015;35(8):801-9. doi: 10.1002/pd.4613.
de la Chapelle A, Herva R, Koivisto M, Aula P. A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet. 1981;57(3):253. doi: 10.1007/BF00278938.
Funato N. Craniofacial phenotypes and genetics of DiGeorge syndrome. J Dev Biol. 2022;10(2):18. doi: 10.3390/jdb10020018.
Sullivan KE. DiGeorge syndrome and chromosome 22q11.2 deletion syndrome. In: Stiehm ER, Ochs HD, Winkelstein JA, editors. Immunologic disorders in infants and children. 5th ed. Philadelphia: Elsevier; 2004. p. 523.
Swillen A, Devriendt K, Vantrappen G, et al. Familial deletions of chromosome 22q11: the Leuven experience. Am J Med Genet. 1998;80(5):531-2.
Hacıhamdioglu B, Hacihamdioglu DO, Delil K. 22q11 deletion syndrome: current perspective. Appl Clin Genet. 2015;8:123-132. doi: 10.2147/TACG.S82105.
Wonkam A, Tokoy R, Cheloy D, Tekendo-Ngongang C, Kingue S, Dahoun S. The 22q11.2 deletion syndrome in congenital heart defects: prevalence of microdeletion syndrome in Cameroon. Glob Heart. 2017;12:115-20. doi: 10.1016/j.gheart.2017.01.003.
Cirillo E, Giardino G, Grasso F, Gallo V, Pignata C. DiGeorge syndrome. In: Rezaei N, editor. Genetic syndromes: a comprehensive reference guide. Cham: Springer; 2022. p. 1-7.https://doi.org/10.1007/978-3-319-66816-1_37-1.
Rumiantsev AG, Mastchan AA. Federal’nye klinicheskie rekomendatsii po diagnostike i lecheniiu sindroma deletsii 22-i khromozomy [Federal clinical recommendations on diagnostics and treatment of the syndrome of 22 chromosome deletion]. Moscow; 2014. 12 p. Russian.
Sgardioli IC, Paoli Monteiro F, Fanti P, Paiva Vieira T, Gil-da-Silva-Lopes VL. Testing criteria for 22q11.2 deletion syndrome: preliminary results of a low cost strategy for public health. Orphanet J Rare Dis. 2019;14(1):123. doi: 10.1186/s13023-019-1098-1.
Siniţîna L, Petrovici V, Tomacinschi C, Şciuca S. Sindromul DiGeorge [DiGeorge syndrome]. Arta Medica (Chisinau). 2017;(4):11-13. Romanian.
Sciuca S, Tomacinschii C, Selevestru R, Sacara V, Maródi L. Inborn errors of immunity in the Republic of Moldova: advances and hope. J Clin Immunol. 2023;43(4):714-716. doi: 10.1007/s10875-023-01439-1.
Kwan A, Puck JM. History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol. 2015;39(3):194. doi: 10.1053/j.semperi.2015.03.004.
Morsheimer M, Whitehorn TFB, Heimall J, Sullivan KE. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am J Med Genet A. 2017;173(9):2366-72. doi: 10.1002/ajmg.a.38319.
Tobias ES, Morrison N, Whiteford ML, Tolmie JL. Towards earlier diagnosis of 22q11 deletions. Arch Dis Child. 1999;81(6):513-514 doi: 10.1136/adc.81.6.513.
Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, Meyts I. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42(7):1508-1520. doi: 10.1007/s10875-022-01352-z.
Chiappini E, Santamaria F, Marseglia GL, Marchisio P, Galli L, Cutrera R, et al. Prevention of recurrent respiratory infections: inter-society Consensus. Ital J Pediatrics. 2021;47(1):211. doi: 10.1186/s13052-021-01150-0.
World Health Organization. Child growth standards. Geneva: WHO; 2025 [cited 2025 Sep 11]. Available from: https://www.who.int/tools/child-growth-standards/standards
Dorif A, Sakara VK, Palii I, Rodoman I, Opalco I, Gladun S. Development of new QF-PCR based digeorge type I syndrome diagnostics method with high prognostic value. In: National Conference with international participation “Natural sciences in the dialogue of generations”; 2023 Sep 14-15; Chisinau, Republic of Moldova: Abstract book. Chisinau: USM; 2023. p. 115.
Digilio MC, Angioni A, De Santis M, Lombardo A, Giannotti A, Dallapiccola B, et al. Spectrum of clinical variability in familial deletion 22q11.2: from full manifestation to extremely mild clinical anomalies. Clin Genet. 2003;63(4):308-13. doi: 10.1034/j.1399-0004.2003.00049.x.
Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34(10):798-804. doi: 10.1136/jmg.34.10.798.
Sullivan KE. The clinical, immunological and molecular spectrum of chromosome 22q11.2.2 deletion syndrome and DiGeorge syndrome. Curr Opin Allergy Clin Immunol. 2004;4(6):505-12, doi: 10.1097/00130832-200412000-00006.
Park I, Ko JK, Kim YH, Yoo HW, Seo EJ, Choi JY, et al. Cardiovascular anomalies in patients with chromosome 22q11.2 deletion: a Korean multicenter study. Int J Cardiol. 2007;114(2):230-5. doi: 10.1016/j.ijcard.2005.12.029.
Jawad AF, McDonald-McGinn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr. 2001;139(5):715-23. doi: 10.1067/mpd.2001.118534.
Oskarsdottir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005;164(3):146-53 doi: 10.1007/s00431-004-1577-8.
Giardino G, Radwan N, Koletsi P, Morrogh DM, Adams S, Ip W, et al. Clinical and immunological features in a cohort of patients with partial DiGeorge syndrome followed at a single center. Blood. 2019;133(24):2586-96. doi: 10.1182/blood.2018885244.
Cancrini C, Puliafito P, Digilio MC, Soresina A, Martino S, Rondelli R, et al. Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J Pediatr. 2014;164(6):1475-80.e2. doi: 10.1016/j.jpeds.2014.01.056.
Nissan E, Katz U, Levy-Shraga Y, Frizinsky S, Carmel E, Gothelf D, Somech R. Clinical features in a large cohort of patients with 22q11.2 deletion syndrome. J Pediatr. 2021;238:215-220.e5. doi: 10.1016/j.jpeds.2021.07.020.
Wonkam A, Tokoy R, Cheloy D, Tekendo-Ngongang C, Kingue S, Dahoun S. The 22q11.2 deletion syndrome in congenital heart defects: prevalence of microdeletion syndrome in Cameroon. Glob Heart. 2017;12(2):115-20. doi: 10.1016/j.gheart.2017.01.003.
Seroogy CM. DiGeorge (22q11.2 deletion) syndrome: clinical features and diagnosis. UpToDate. Updated 2019 June 11. Accessed 2025 Sep 1.
Rosa RF, Rosa, RC, Dos Santos PP, Zen PR, Paskulin GA. Hematological abnormalities and 22q11.2 deletion syndrome. Rev Bras Hematol Hemoter. 2011;33(2):151-154. doi: 10.5581/1516-8484.20110037.
Biggs SE, Gilchrist B, May KR. Chromosome 22q11.2 deletion (DiGeorge syndrome): immunologic features, diagnosis, and management. Curr Allergy Asthma Rep. 2023;23(4):213-222. doi: 10.1007/s11882-023-01071-4.
Sullivan KE. Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunol Rev. 2019;287(1):186-201. doi: 10.1111/imr.12701.